

Neuromuscular Disorders
Viral and retroviral myositis
Jun. 16, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Disorders of lipid metabolism are a heterogeneous group of diseases that cause excessive lipid storage in multiple organs, with skeletal and cardiac muscle most commonly affected. Although rare, definitive diagnosis is crucial as treatment with specific supplements or dietary modification can improve clinical outcomes in some patients. The main clinical phenotypes consist of fixed progressive muscle weakness or episodic rhabdomyolysis with variable cardiomyopathy. These disorders are mostly genetic in origin and are the main subject of this review. Acquired causes of lipid myopathy are briefly addressed.
|
• Lipid myopathies are genetic disorders with autosomal recessive transmission. Therefore, they have no significant gender predominance and are more common in children of consanguineous parents. | |
|
• Classic lipid myopathies with massive lipid accumulation tend to present with fixed and progressive muscle weakness and cardiomyopathy. | |
|
• Lipid disorders with modest tissue lipid deposits tend to cause a phenotype of episodic weakness with rhabdomyolysis triggered by metabolic stress. | |
|
• LPIN1 and carnitine palmitoyltransferase II deficiencies are the most common genetic causes of recurrent rhabdomyolysis in children and adults, respectively. | |
|
• In all lipid myopathies, measurements of plasma acylcarnitines, urinary organic acid profile, and detection of the Jordan anomaly in a peripheral blood smear can be useful diagnostic tools. Genetic testing or enzymatic assays can then provide a definitive diagnosis. | |
|
• Treatment involves avoiding triggers of metabolic stress, supplementation (carnitine, riboflavin, and coenzyme Q10), and dietary modifications. |
In 1953, Jordan first described lipid vacuoles in leucocytes of two brothers with “dystrophia musculorum progressive.” This finding (Jordan anomaly) in the peripheral blood smears of patients with neutral lipid storage disease is pathognomonic and a useful tool for diagnosis. Maurice Dorfman first reported a case of neutral lipid storage disease with skin phenotype (“ichthyosiform dermatosis”) in 1974; Chanarin confirmed this phenotype in 1975.
Engel first reported episodic myalgias and rhabdomyolysis secondary to a long-chain fatty acid metabolism deficit in 1970. Carnitine palmitoyltransferase II deficiency, the most common genetic cause of recurrent rhabdomyolysis, was the first muscle fatty acid oxidation disorder to be identified (20). In 1974, Engel and Angelini reported carnitine deficiency as a cause of lipid storage myopathy.
Lipid metabolic myopathies can result from deficits at different stages:
|
• Intracellular triglyceride synthesis or catabolism | |
|
• Transport of long-chain fatty acids and carnitine system | |
|
• Beta-oxidation |
Lipid myopathies are characterized by variable lipid accumulation in muscle and in other tissues and two main phenotypes of energy failure: (1) fixed or progressive muscle weakness, with or without metabolic crisis, seen in classic lipid storage myopathies; and (2) recurrent rhabdomyolysis triggered by fasting, infection, or prolonged exercise typical of mitochondrial fatty acid transport or beta-oxidation defects.
Clinical presentation. There is a wide spectrum of clinical manifestations, but this condition mostly affects the liver and skeletal and cardiac muscle. The infantile-onset form manifests with attacks of hypoglycemic hypoketotic encephalopathy with hyperammonemia (Reye-like syndrome) triggered by fasting or concurrent infection. In the childhood-onset form, usually with onset at 4 years of age, predominant symptoms are progressive hypertrophic or dilated cardiomyopathy and proximal muscle weakness with elevated creatine kinase. Heart failure develops by 10 years of age unless carnitine supplementation is started early (01). A rare adult-onset form manifests with easy fatigability, myalgias, or, rarely, cardiomyopathy. Women may develop these types of symptoms during pregnancy, which unmasks the disease (23).
Carnitine deficiency restricted to skeletal muscle (OMIM #212160) has been reported (24). There is a wide clinical spectrum, and the age of onset ranges from childhood to adulthood. Clinical features include fluctuating muscle weakness, axial myopathy with hypoventilation, and masticatory weakness (115; 43). Muscle carnitine levels are low (< 15%), but contrary to the systemic form, plasma levels are normal and urinary levels are not increased.
Etiology and pathogenesis. Carnitine, an amino acid derivative, is an essential cofactor in the beta-oxidation of fatty acids, and its sources include diet (animal products) and de novo synthesis. Carnitine deficiency due to reduced dietary intake is extremely rare. Even in the context of malnutrition or strict vegetarianism, biosynthesis and increased renal reabsorption can compensate for reduced intake (58).
Primary carnitine deficiency is an autosomal recessive disorder with a frequency of about one in 100,000 newborns; the highest prevalence noted is in the Faroe Islands, where it reaches one in 3000 births (60).
There is a defect in the organic cation transporter type 2, which is encoded by the SLC22A5 gene and transports carnitine across the plasma membrane. The transporter residual activity seems to correlate with the severity of disease and age of onset (87). Heterozygous carriers are asymptomatic and do not seem to be at risk of cardiomyopathy (03).
The low activity of the organic cation transporter in the kidneys results in urinary carnitine wasting, with secondary low serum and tissue intracellular concentration. Carnitine is required for the transfer of long-chain fatty acids from the cytosol to the mitochondria, where beta-oxidation occurs. Carnitine deficiency has two major consequences: (1) defective fatty acid oxidation resulting in hypoglycemia due to excessive glucose consumption without regeneration via gluconeogenesis; and (2) the inability to use fat released from adipose tissue during fasting, resulting in intracellular lipid accumulation in the liver and skeletal and cardiac muscle with secondary cellular dysfunction.
In the muscle-restricted form of carnitine deficiency, there is decreased cellular uptake despite normal systemic and urinary concentrations (115).
Evaluation and diagnosis. Muscle biopsy typically reveals a massive lipidosis with vacuoles affecting mostly type 1 fibers and type 2 fibers, demonstrating atrophy. Creatine kinase is moderately elevated (3x to 5x). Plasma-free carnitine levels are severely reduced (less than 5 uM, normal 25 to 50 uM). The acylcarnitine profile is normal or shows reduction in species, which is helpful to differentiate primary carnitine deficiency from secondary carnitine reduction due to long-chain oxidation defects (carnitine palmitoyltransferase deficiency, very long-chain acyl-CoA dehydrogenase deficiency).
Genetic testing demonstrates homozygous mutations in the SLC22A5 gene. Carnitine uptake reduction can be demonstrated on skin fibroblasts or lymphocytes, with a reduction of up to 10% of controls (50). This latter test is especially helpful for confirmation if genetic testing is negative but clinical suspicion remains high.
In neonates with positive screening, maternal history and carnitine levels can be informative. Carnitine crosses the placenta, so neonates can show low levels due to primary disease or secondary-to-low maternal carnitine levels. In the latter case, neonate levels tend to normalize after a few weeks.
Treatment and management. High-dose oral carnitine (100 to 400 mg/kg/day) in three to four daily doses provides dramatic improvement in symptoms and ameliorates or prevents the development of cardiomyopathy (84; 50). The dose needs to be titrated to maintain normal plasma levels, and supplementation should be lifelong (01). Newborns are routinely screened for primary carnitine deficiency by dried blood spot testing, and supplementation is started as soon as the condition is suspected. Side effects of L-carnitine include diarrhea and fishy body odor (production of trimethylamine). Overall, long-term prognosis is good if individuals remain on carnitine supplementation.
It is important to avoid drugs that can worsen or cause primary carnitine deficiency to flare. Pivolic acid is a compound used to increase the bioavailability of certain antibiotics (pivampicillin, pivmecillinam, etc). It can inactivate carnitine by esterification and acutely deteriorate patients with this condition (81). Valproic acid can also lower carnitine levels by reducing biosynthesis and increasing excretion (53).
Neutral lipid storage diseases are rare autosomal recessive disorders characterized by nonlysosomal accumulation of triglycerides in the form of cytoplasmic lipid droplets affecting multiple tissues.
Clinical presentation. Neutral lipid storage disorder with ichthyosis presents earlier in life compared to neutral lipid storage disorder with myopathy (22). Ichthyosis (nonbullous congenital ichthyosiform erythroderma) manifests as dry, scaly skin and affects most of the body’s surface; it is always present in childhood. More variable features include short stature, microcephaly, mental retardation, hepatosplenomegaly, ophthalmic pathology (cataracts, strabismus), and intestinal involvement (21; 52). A lipid myopathy, which typically begins in the second or third decade of life, is present in up to 40% of cases (82). Unlike neutral lipid storage disorder with myopathy, cardiomyopathy is not common. Fatty liver can eventually cause liver failure and can be fatal (78).
Etiology and pathogenesis. Neutral lipid storage disorder with ichthyosis is caused by a defect in the ABHD5 gene, which codes for comparative gene identification #58 (CGI-58), an activator of adipose triglyceride lipase. This enzyme is responsible for the first and rate-limiting step in lipolysis that releases the first fatty acid from the glycerol backbone. Therefore, dysfunction of adipose triglyceride lipase affects degradation of intracellular triglycerides, leading to its accumulation. Inheritance is autosomal recessive, and most cases seem to originate in Middle Eastern countries around the Mediterranean basin (52).
Clinical presentation. The main manifestations include myalgia and a gradually progressive myopathy, which are usually noticeable by the third decade of life (83). The pattern of weakness is variable: proximal and distal, distal predominant, or axial myopathy with neck extensor involvement (dropped head syndrome) have been described (71; 70; 25; 51). Asymmetry and upper limb predominance, usually worse on the right side, can be present at onset or later in the course (78; 96; 09). Dilated cardiomyopathy leading to heart failure and conduction abnormalities is a frequent complication (approximately 50%), although the severity is quite variable (83). Respiratory muscle involvement is not a common feature. Other variable systemic manifestations include liver impairment, sensorineural hearing loss, pancreatitis, and diabetes (67; 51).
A clear genotype-phenotype correlation has not been found; furthermore, the same mutation can cause significant clinical heterogeneity within the same family (65).
Etiology and pathogenesis. There is an autosomal recessive defect in the patatin-like phospholipase domain containing 2 (PNPLA2) gene, which codes for the protein adipose triglyceride lipase. This enzyme catalyzes the first and rate-limiting step of lipid hydrolysis in the cytoplasm, converting triacylglycerol to diacylglycerol and a free fatty acid. With this enzymatic deficit, normal triacylglycerol accumulates excessively in the cells of several tissues.
Evaluation and diagnosis (neutral lipid storage disorder with ichthyosis and neutral lipid storage disorder with myopathy). The most characteristic laboratory abnormality in neutral lipid storage diseases is the multiple lipid inclusions inside neutrophils and eosinophils (Jordan anomaly), which are visible in the peripheral blood smear in 100% of cases (40; 78). The percentage of leukocytes with lipid droplets varies from 10% to 100%, and this finding can also be present in marrow megakaryocytes.
Creatine kinase is usually elevated (average 1000 IU/L), especially in the neutral lipid storage disorder with myopathy form (96). Serum triglycerides are typically elevated, but serum carnitine and plasma acylcarnitine profiles are normal. EMG studies typically show a myopathic or mixed (myopathic and neurogenic) pattern with myotonic discharges (78). Muscle MRI may show fatty replacement of posterior leg and thigh muscles (96).
Lipid accumulation in muscle fibers (neutral lipid storage disorder with myopathy), liver cells, or skin keratinocytes (neutral lipid storage disorder with ichthyosis) can be helpful for diagnosis. Muscle biopsy typically reveals nonrimmed vacuolization of fibers without significant connective tissue deposition, even in patients without significant muscle weakness. Chronic myopathic changes (fiber size variation, internalization of nuclei) without inflammation or significant necrosis are detected. Lipid droplets can be sarcoplasmic or subsarcolemmal and are easily visible by optic microscopy (“Swiss cheese appearance”). They are strongly positive for Oil Red O and Sudan Black staining (78; 25; 51) and predominantly affect type 1 muscle fibers. Of note, rimmed vacuoles can be present in some neutral lipid storage disorder with myopathy patients, suggesting a pathomechanism beyond lipid accumulation (70; 96).
Genetic confirmation is achieved by the analysis of ABHD5 (neutral lipid storage disorder with ichthyosis) or PNPLA2 (neutral lipid storage disorder with myopathy) genes, which reveal homozygous mutations.
Treatment and management. There is no specific treatment available. Urea containing topical emollients can be helpful for ichthyosis. The dietary regimen should focus on a low-fat diet with medium-chain triglyceride supplementation and an increase in carbohydrates as an alternative source of energy (48). Bezafibrate demonstrated experimental, but not clinical, evidence of efficacy (112). Cardiomyopathy in neutral lipid storage disorder with myopathy can be fatal and may require a pacemaker, defibrillator, or transplantation (54).
Clinical presentation. The neonatal form, with (type I) or without (type II) congenital anomalies, results from complete enzyme deficiency and is almost universally fatal in the first few days or weeks of life due to cystic renal dysplasia, liver dysfunction, and hypoglycemia (32). The late-onset or milder form (type III) is characterized by myopathy and episodic metabolic acidosis, vomiting, and hepatomegaly (33). This condition was the most the common genetic etiology (44.6%) in a large cohort of rhabdomyolysis (08). A systematic review found male predominance (58%) in the late-onset form (61).
Onset in childhood or adolescence is characterized by a mix of myopathic symptoms and repeated episodes of hypoglycemia, acidosis, and liver dysfunction. On the contrary, onset in adulthood is predominantly characterized by skeletal muscle-related symptoms without systemic involvement (07).
Muscle disease in late-onset multiple acyl-CoA dehydrogenase deficiency includes a myopathy associated with myalgia, fatigue, and occasional rhabdomyolysis. Fixed myopathy is the most common pattern (85%), followed by acute muscular symptoms in one third of cases and a mix of both in 20% (33). Myopathy is typically proximal and axial, with involvement of neck extensors (“dropped head syndrome”), paraspinal muscle atrophy, and masticatory weakness (56; 123; 114; 119). Symptoms in the neck and bulbar muscles can have “pseudo-myasthenic” fluctuations and can worsen during metabolic stress (27; 07). The subacute course of proximal myopathy and high creatine kinase can sometimes mimic polymyositis (120; 92).
Intermittent or cyclic metabolic deterioration (encephalopathy, vomiting, hypoglycemia, and acidosis) in the context of fasting, surgery, or concurrent febrile illness can be life-threatening (56; 07). Like primary carnitine deficiency, the disease can also be unmasked during pregnancy (37). Extramuscular manifestations include fatty liver or “transaminitis” sometimes associated with delirium. Of note, cardiomyopathy is not a prominent feature of this disorder.
The allelic disorder coenzyme Q10 deficiency predominantly manifests with muscle symptoms, such as cases of late-onset multiple acyl-CoA dehydrogenase deficiency without coenzyme Q10 deficiency (37; 27). Patients present a phenotype of proximal myopathy, fatigue, and exercise intolerance without the CNS manifestations (ataxia, epilepsy, encephalopathy) seen in other types of primary coenzyme Q10 deficiency (98).
Etiology and pathogenesis. This is an autosomal recessive disorder due to mutations in three different genes encoding for electron-transferring flavoproteins (ETFA, ETFB) and flavoprotein dehydrogenase (ETFDH). These proteins contain flavin adenine dinucleotide prosthetic groups, and flavin adenine dinucleotide levels could affect the folding and maintenance of their native structure (93). The flavin dependence of these proteins likely explains the beneficial effect of riboflavin supplementation (28).
Electron-transferring flavoproteins are localized in the mitochondrial matrix and receive electrons from acyl-CoA dehydrogenases involved in fatty acid oxidation. ETFDH resides in the inner mitochondrial membrane, receives electrons from electron-transferring flavoproteins, and transfers them to the respiratory chain (116). Defective electron-transferring flavoproteins disturb the electron transfer and result in the accumulation of acyl-CoA esters, decrease of intra-mitochondrial flavin, and mitochondrial dysfunction.
Mutations in the ETFA and ETFB genes are responsible for the fatal neonatal form. Less severe mutations of the ETFDH gene, which result in some residual enzymatic activity, are responsible for most cases of late-onset multiple acyl-CoA dehydrogenase deficiency (74) and the myopathic form of coenzyme Q10 deficiency (73; 27). Carriers of heterozygous pathogenic mutations may develop a MADD-like subacute myopathy when exposed to the sertraline antidepressant, possibly through specific mitochondrial toxicity (117).
The mechanism for coenzyme Q10 deficiency remains unclear. It has been suggested that reduction in the dehydrogenase (due to ETFDH mutation) could downregulate the synthesis of coenzyme Q10 via a feedback mechanism (27). Alternatively, faulty binding of the dehydrogenase to coenzyme Q10 could result in degradation of the electron acceptor molecule (73).
Evaluation and diagnosis. Muscle biopsy shows a typical lipid storage myopathy with mitochondrial changes (occasional ragged-red fibers, COX-negative fibers). Muscle fibers with cracks on hematoxylin and eosin and decreased succinate dehydrogenase (SDH) activity can help differentiate MADD from other lipid myopathies (119). Muscle biopsy may be more useful in cases of very late onset (91). Liver biopsy may also show “foamy” hepatocytes suggestive of a lipid storage disorder (56). Baseline creatine kinase is moderately elevated (up to 2000 IU/L of 1512 IU/L) outside episodes of rhabdomyolysis (119).
Plasma carnitine concentration can be reduced (secondary reduction), but not as severely as in primary carnitine deficiency. Acylcarnitine profile is paramount for diagnosis as it shows a characteristic increase of short-, medium-, and long-chain species (C4 to C18:1). Urinary organic acid analysis can be abnormal, with the presence of dicarboxylic aciduria (C5-C10, glutaric acid) and acylglycine derivates. Testing of these metabolic abnormalities in plasma and urine has a higher yield during an acute exacerbation or fasting state (12 hours or longer).
Coenzyme Q10 normally transfers electrons from complexes I and II to complex III of the mitochondrial respiratory chain. Myopathic coenzyme Q10 deficiency shows lipid storage myopathy and decreased coenzyme Q10 in muscle tissue. Respiratory chain dysfunction is a hallmark of the disease, with reduction in the activity of mitochondrial complexes (I to IV). Serum creatine kinase and lactate are typically elevated (37; 27).
Genetic confirmation is achieved by finding recessive mutations (homozygous or compound heterozygous) in the ETFA, ETFB, or ETFDH genes. The latter one is responsible for most cases of early- and late-onset (type III) multiple acyl-CoA dehydrogenase deficiency, but no clear genotype-phenotype has been found (07).
Treatment and management. Late-onset multiple acyl-CoA dehydrogenase deficiency (type III) is a treatable condition, so prompt recognition is key. Riboflavin is a water-soluble vitamin (B2) with rare toxicity. Riboflavin (50-100 mg three times daily) produces a dramatic improvement of symptoms in the chronic form of the disease but can also rescue acute exacerbations (74; 36; 118; 57; 119). Adding carnitine to riboflavin is logical (given the secondary reduction) and has been reported as beneficial in late-onset multiple acyl-CoA dehydrogenase deficiency (56; 07). However, carnitine may be ineffective or even cause clinical worsening in cases with secondary coenzyme Q10 deficiency (27). Patients with the myopathic form of coenzyme Q10 deficiency showed significant improvement with combined coenzyme Q10 (150-500 mg/day) and riboflavin supplementation (37; 27).
Misdiagnosis of polymyositis can lead to treatment with steroids, but no clinical benefit is typically seen (119). Oral steroids have proved to be ineffective in multiple acyl-CoA dehydrogenase deficiency when compared to riboflavin (57).
Prevention of attacks by avoiding triggers is key. Besides replacement therapy, a low-fat diet and avoidance of fasting can prevent a metabolic crisis (36; 120).
Other subsets of riboflavin-responsive disorders present similarly to multiple acyl-CoA dehydrogenase deficiency, but mutations affect different genes, including the riboflavin transporter, flavin adenine dinucleotide transporter, or flavin adenine dinucleotide synthase (94; 06).
Clinical presentation. This condition is one of the most common fatty acid oxidation defects, but skeletal or cardiac muscle impairment is rare. Onset is in infancy with episodic hypoketotic hypoglycemia and Reye-like syndrome with hyperammonemia triggered by fasting or febrile illness. Acute metabolic crises in early life cause high morbidity and mortality rates due to potential liver failure, apnea, and seizures (17).
Some patients with a milder form develop myopathy in the second decade of life, and a limb-girdle pattern of weakness with dropped head syndrome has been described (114). Chronic exercise intolerance with myalgias can be present in around one third of patients (17). Patients with the late-onset form are also at risk for episodic rhabdomyolysis and acute metabolic deterioration triggered by alcohol or fasting (90; 17).
Etiology and pathogenesis. Medium-chain acyl-CoA dehydrogenase deficiency is essential for the oxidation of activated fatty acids of C4 to C12 carbon atom length.
Evaluation and diagnosis. The acylcarnitine profile shows a characteristic increase in medium-chain species (C6, C8, C10, C10:1), which is included in newborn screening. Corresponding free fatty acids (octanoic, decenoic) are increased in blood and urine samples. Dicarboxylic aciduria is also present. Secondary carnitine deficiency is common. In cases of myopathy, lipid excess can be demonstrated in the muscle biopsy. Reduced enzymatic activity (< 10%) can be demonstrated in muscle, fibroblasts, or liver tissue. Genetic testing confirms the condition with homozygous mutations in the ACADM gene (acyl-CoA dehydrogenase medium-chain), with C.985A>G (p.Lys304gluc) being the most common alteration.
Treatment and management. There is no specific treatment other than the avoidance of triggers. Carnitine supplementation was suggested in the past but was later found to be ineffective in controlled studies (38; 62).
Clinical presentation. Clinical heterogeneity is prominent in this disorder, including asymptomatic patients and features of central nervous system and neuromuscular impairment. Patients commonly present in early childhood with nonspecific symptomatology, including developmental delay, acidosis, hypotonia, behavioral disorders, and epilepsy. Contrary to medium-chain acyl-CoA dehydrogenase deficiency and very long-chain acyl-CoA dehydrogenase deficiency, hypoglycemia or acute metabolic decompensations are not frequent in this disorder (104). Late-onset phenotypes are infrequent and are characterized by subtle progressive proximal myopathy (104). A peculiar variant characterized by progressive external ophthalmoplegia and ptosis with scoliosis, contractures, cardiomyopathy, and multicore myopathy has been described (105; 104).
Etiology and pathogenesis. The estimated prevalence is one in 50,000 (113). This disorder is related to a deficiency of short-chain acyl-CoA dehydrogenase (111). This enzyme, which is located in the mitochondrial matrix, is involved in short-chain (four to eight carbons in length) fatty acid beta-oxidation. Genetic and environmental factors likely play a role in this disorder due to reduced penetrance. Furthermore, in benign variants of the ACADS gene, disease susceptibility alleles and causative mutations are difficult to differentiate from each other (104).
Evaluation and diagnosis. A laboratory hallmark is an increase in urinary ethylmalonic acid and plasma acylcarnitine profile, which demonstrates an increase in butyrylcarnitine (C4). Muscle histology may reveal either lipid storage myopathy or multiminicore features (111; 104). Diagnostic confirmation is obtained by confirmation of homozygous mutations in the ACADS gene.
Treatment and management. There is no specific treatment. Avoidance of fasting with low-fat and high-carbohydrate diets is recommended (104).
Differential diagnosis. To date, four lipid storage myopathies have been described: primary carnitine deficiency, neutral lipid storage disorder with ichthyosis, neutral lipid storage disorder with myopathy, and multiple acyl-CoA dehydrogenase deficiency. Beyond the clinical differences, the acylcarnitine profile is normal in primary carnitine deficiency and neutral lipid storage disease, whereas in multiple acyl-CoA dehydrogenase deficiency, there is an elevation of all length-chain carnitines, and urine organic acid analysis reveals dicarboxylic acids and acylglycine derivatives.
Primary carnitine deficiency should be differentiated from secondary causes of carnitine deficiency, which include genetic and acquired causes. Among the former ones, fatty oxidation defects (very long-chain acyl-CoA dehydrogenase deficiency, medium-chain acyl-CoA dehydrogenase deficiency, and carnitine palmitoyltransferase II deficiency) should be considered where the carnitine deficit is moderate (5-25 uM) and acylcarnitine profiles demonstrate abnormalities. On the contrary, primary carnitine deficiency is characterized by a very low level of plasma carnitine (< 5 uM), with a normal acylcarnitine profile. Acquired causes of carnitine deficiency include malnutrition, total parental nutrition, renal tubular dysfunction, and medications (cyclosporine, valproate, and pivampicillin). Removal of the offending agent should improve plasma levels in these secondary cases. Supplementation should be done if it is not possible to remove the agent.
Multiple acyl-CoA dehydrogenase deficiency and neutral lipid storage disorder with myopathy can initially be mistaken for polymyositis given the common presentation of proximal weakness and elevated creatine kinase (25; 92). Muscle biopsy is helpful for the differentiation of these conditions by demonstrating a lipid storage myopathy and lack of inflammation. Treatment with steroids is not only ineffective but can worsen lipid myopathies, and this can be a clue to an alternative diagnosis (57).
Multiple acyl-CoA dehydrogenase deficiency causes mitochondrial changes on muscle biopsy and secondary reduction in respiratory chain complex activity, which can be difficult to distinguish from primary mitochondrial respiratory chain disorders. However, the overt lipid storage in type 1 fibers of multiple acyl-CoA dehydrogenase deficiency is not present in classic mitochondrial disorders.
Other important differential diagnoses of lipid storage myopathies include muscular dystrophies (eg, limb-girdle muscular dystrophy, facioscapulohumeral muscular dystrophy) and adult-onset Pompe disease, for which the muscle biopsy is paramount in making the distinction. Type IV spinal muscular atrophy can have a similar presentation of proximal weakness, but there are commonly fasciculations, hyporeflexia, or areflexia, and a chronic neurogenic process (rather than myopathic) is present on EMG.
The short-chain acyl-CoA dehydrogenase deficiency variant with chronic progressive ophthalmoplegia, ptosis, and multicore myopathy should be differentiated from mitochondrial myopathies or congenital myopathies with RYR1 or SEP1 mutations.
Skeletal and cardiac muscles are highly oxidative tissues that utilize fatty acid substrates as a source of energy. The liver is the other organ that metabolizes fatty acids. Therefore, skeletal and cardiac muscle and the liver are the main organs that are impaired in fatty acid oxidation disorders.
Fatty acids are the main source of muscle energy at rest during prolonged (> 10 minutes) or low-intensity exercise. In the initial phases of slow exercise or during quick strenuous exercise, carbohydrates (glycogen) are the main source of adenosine triphosphate generation in skeletal muscle. When the energy demand increases and exceeds what is generated by glycolysis, there is a switch to fatty acid metabolism as the main source of energy. The beta-oxidation pathway is also enhanced during fasting or febrile states (99). If the adenosine triphosphate generated by lipid metabolism cannot keep up with the energy demands, muscle symptoms will develop.
Depending on the age of onset and severity, fatty acid oxidation disorders generally have three forms of clinical presentation. A neonatal form with predominant hypoglycemia, encephalopathy, and cardiomyopathy is most often fatal. An infantile form has similar, but less severe, episodic symptoms. A milder late-onset form has predominant muscle symptoms, including exercise intolerance and rhabdomyolysis.
Treatment consists of the reduction or modification of fatty acid content and an increase in carbohydrate intake. Given the relative benefit of this treatment, fatty acid oxidation disorders have been included in newborn screening, with acylcarnitine assay. Proper diagnosis and management have been associated with reduction in admission to intensive care unit and frequency of rhabdomyolysis (88).
Clinical presentation. Three main clinical phenotypes have been described: (1) a severe neonatal form with cardiomyopathy, cardiac arrhythmias, liver failure, and risk of sudden death; (2) an intermediate childhood-onset form with episodic hypoketotic hypoglycemia and hepatomegaly triggered by fasting or febrile illness; and (3) a juvenile or adult-onset form characterized mainly by recurrent rhabdomyolysis and exercise intolerance (69; 04; 47).
Skeletal muscle symptoms typically predominate the clinical picture after the age of 10 (04; 47). Rhabdomyolysis can be triggered by prolonged exercise, febrile illness, and exposure to cold or prolonged fasting. A combination of factors as a trigger is common, such as exercise and exposure to cold with winter sports. The frequency of these episodes varies significantly, from less than one to six attacks per year. Muscle cramps and exercise intolerance without overt rhabdomyolysis are also common.
Although rare, metabolic crises have been reported in juvenile or adult patients with very long-chain acyl-CoA dehydrogenase deficiency. Complications include acute cardiac failure, hypercapnic respiratory failure, metabolic acidosis, and persistent hypoglycemia (46; 109; 31).
A percentage of patients remain asymptomatic when diagnosed by newborn screening and observed longitudinally (100).
Etiology and pathogenesis. Very long-chain acyl-CoA dehydrogenase deficiency is an autosomal recessive disorder due to mutations in the ACADVL gene. According to newborn screening, it affects one in 63,481 births in the United States (107).
Very long-chain acyl-CoA dehydrogenase is an enzyme located in the inner mitochondrial membrane that catalyzes the initial step in the beta-oxidation of fatty acids with very long-chain length (14-20 carbons), which are converted in acetyl-CoA and ketones (39). Very long-chain acyl-CoA dehydrogenase is immediately downstream from carnitine palmitoyltransferase II, as the latter one incorporates long-chain fatty acids into the mitochondrial matrix.
When very long-chain acyl-CoA dehydrogenase is deficient, there is an inability to metabolize fatty acids at times of increased metabolic requirements. The residual enzymatic activity in the late-onset form can be overwhelmed during prolonged exercise, concurrent illness, or fasting. The resulting accumulation of fatty acid intermediates may have a deleterious, toxic, detergent-like effect on the muscle membranes, thereby causing muscular symptoms and rhabdomyolysis.
Evaluation and diagnosis. Baseline creatine kinase levels are usually normal or moderately elevated but will rise significantly during metabolic stress (89). Muscle biopsy shows mild lipid accumulation in approximately a third of the cases (47) but is uninformative in most cases. Low free serum carnitine levels (secondary reduction) can be a clue to a fatty acid oxidation disorder. MRI T1W sequences can show nonspecific signal changes in proximal muscles, reflecting lipid accumulation (19).
The most important biochemical test is the plasma acylcarnitine profile, which reveals elevated C14 long-chain species (C14:1, C14:2m, and C14), and the yield is increased if testing is performed during fasting or rhabdomyolysis. A urinary organic acid profile can show dicarboxylic aciduria at the time of acute attacks. Diagnostic confirmation is achieved by demonstrating reduced enzymatic activity in skin fibroblasts or lymphocytes (< 12%) and by molecular analysis of the ACADVL gene, demonstrating biallelic mutation.
Genotype-phenotype correlation is present in very long-chain acyl-CoA dehydrogenase deficiency. Severe early-onset disease is associated with no residual enzyme activity and null mutations. Milder late-onset forms are usually due to variants, resulting in residual enzymatic activity (04; 18).
Treatment and management. There is no proven effective treatment for very long-chain acyl-CoA dehydrogenase deficiency. The avoidance of triggers (fasting, cold) is paramount. A low-fat diet (reduction in long-chain fatty acids) with medium-chain triglycerides and triheptanoin supplementation is possibly effective in reducing rhabdomyolysis and improving cardiomyopathy in the childhood-onset form (85; 30). The rationale for medium-chain triglyceride supplementation is that medium-chain triglycerides bypass the carnitine shuttle, and there is a reduction in the production of toxic long-chain esters by conversion into ketone bodies. Medium-branched-chain fatty acids improved the mitochondrial energy profile of fibroblasts from patients with long and very-long-chain fatty acid oxidation disorders (44). Despite dietary modifications, a third of patients will remain symptomatic (100).
Intravenous glucose and oral medium-chain triglyceride supplementation were not effective in improving exercise intolerance in the adult-onset form of very long-chain acyl-CoA dehydrogenase deficiency (76). Bezafibrate and resveratrol were studied in randomized, double-blind crossover trials, but no clinical benefit was demonstrated for either compound (75; 101). Finally, in patients with secondary carnitine deficiency, supplementation is typically implemented but remains controversial (89).
Life-threatening events and extramuscular manifestations (like nonketotic hypoglycemia) tend to happen early in life; long-term studies have shown that if patients overcome the first few years, the prognosis is quite favorable (47).
Experimental therapy with a single intravenous injection of adenovirus-associated vector 9 (AAV9)-mediated very long-chain acyl-CoA dehydrogenase gene replacement has been tried in an experimental mouse model (125). This promising treatment ameliorated lipid accumulation and respiratory weakness in fasting mice.
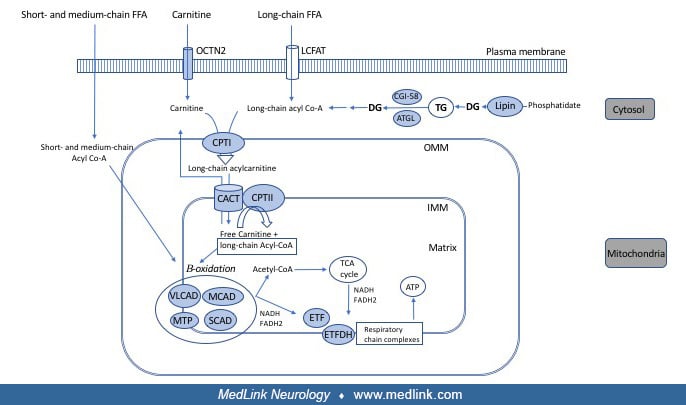
Long-chain fatty acids cannot freely diffuse into the cell. They utilize a cell membrane-specific transporter, the long-chain fatty acid translocase, to get into the cytosol. The mitochondrial membrane also has selective permeability, and the carnitine-acylcarnitine transport mechanism is necessary to transport long-chain fatty acids into the mitochondrial matrix, where they undergo beta-oxidation. Fatty acids are first activated by long-chain acyl-CoA synthetase in the outer mitochondrial membrane. Long-chain acyl-CoAs are the substrate of carnitine palmitoyltransferase I, which catalyzes the formation of acylcarnitine complex (ie, fatty acid + carnitine); this complex is then transported inside the mitochondria via carnitine-acylcarnitine translocase. In the inner mitochondrial membrane, carnitine palmitoyltransferase II catalyzes separation of the complex, resulting in the formation of acyl-CoA and free carnitine. The latter is available to restart this cycle of transport, and acyl-CoA undergoes beta-oxidation in the mitochondrial matrix.
Carnitine palmitoyltransferase II and carnitine-acylcarnitine translocase deficiencies are further discussed in this article. Carnitine palmitoyltransferase I deficiency causes selective liver involvement without neuromuscular symptomatology.
Clinical presentation. There are three clinical phenotypes depending on the severity and age of onset. A lethal neonatal form and the severe infantile forms are characterized by systemic involvement with liver failure, hypoketotic hypoglycemia, cardiomyopathy, seizures, and sudden death. Milder enzyme deficiency allows for survival into adulthood and causes the classic myopathic form, which is the most common disorder of lipid metabolism affecting skeletal muscle.
Despite being an autosomal recessive disorder, a clear male predominance (3:1) is seen in published cases. Ascertainment bias, given that males are more likely to engage in strenuous exercise and the influence of estrogen in carnitine palmitoyltransferase activity regulation, has been suggested as a plausible explanation for this gender distribution (05; 77).
Although referred to as “adult” onset, the classic myopathic form can start from childhood into the early adulthood years (121; 41). Symptoms mostly consist of exercise intolerance, with myalgias, cramps, and muscle stiffness or contractures. Recurrent rhabdomyolysis and myoglobinuria is frequent in this condition (20; 121). Attacks can be triggered by fasting, a high-fat diet, general anesthesia, prolonged exercise, and exposure to cold or febrile illness. Of all of these factors, prolonged exercise (> 15 minutes) is the most common trigger (41; 95). Rhabdomyolysis with onset 5 hours post Covid-19 vaccination has been reported (103). The frequency of attacks can vary from one to 85 per year, with an average of 14 attacks (42).
Most patients develop attacks starting in childhood or in the teenage years. In a case series of 13 patients, the age of the first attack ranged from 4 to 18 years, with a mean age of 9.7 years (42). Affected muscles include limb musculature (predominantly legs), but facial or jaw muscle involvement is not unusual (42).
In 90% of cases, muscle strength and creatine kinase levels are normal between episodes (108; 121). Fixed muscle weakness is very rare, and cardiomyopathy is not a feature of the myopathic form. The main extramuscular manifestation is potential kidney injury during rhabdomyolysis episodes, which can rarely lead to permanent nephropathy and the need for dialysis.
Etiology and pathogenesis. Carnitine palmitoyltransferase II deficiency leads to an inability to utilize long-chain fatty acids for adenosine triphosphate generation. Total carnitine palmitoyltransferase concentration in muscle tissue is comparable to controls, but there is abnormal residual carnitine palmitoyltransferase II activity with the addition of malonyl-CoA and Triton X-100 (41). This suggests that the pathogenesis is not exclusively due to a reduction in enzyme activity but also to abnormal enzymatic regulation during situations that require increased fatty acid metabolism. The thermolability of this enzyme seen in experimental models could explain febrile illness as a common trigger for attacks in carnitine palmitoyltransferase II deficiency (66).
Evaluation and diagnosis. The reduction in the conversion of acylcarnitine into acyl-CoA and free carnitine results in a plasma accumulation of acylcarnitines that can be measured for diagnostic purposes. The plasma acylcarnitine profile shows an increase in C16 and C18 species, and yield is increased if measured during a fasting state or rhabdomyolysis. An increased ratio of (C16:0+C18:1)/C2>0.62 is reflective of these abnormalities (26). There is normal or mild secondary reduction in plasma carnitine levels. The acylcarnitine profile abnormalities are like those present in very long-chain acyl-CoA dehydrogenase or carnitine-acylcarnitine translocase deficiencies, but the lack of dicarboxylic or glutaric aciduria in carnitine palmitoyltransferase II can be a helpful differentiating feature.
Muscle biopsy is usually normal or shows mild, nonspecific myopathic findings. A mild muscle lipidosis is present in about 20% of patients (49). During attacks, isolated necrotic fibers can be present.
Carnitine palmitoyltransferase II enzyme activity reduction can be demonstrated in blood lymphocytes and skin fibroblast. Residual carnitine palmitoyltransferase II activity reduction can also be demonstrated in muscle tissue.
Confirmation is best achieved by genetic testing, with the demonstration of one homozygous or two compound-heterozygous mutations. The most prevalent mutation identified in the adult myopathic form is C338C>T, p.Ser113Leu, which is present in 70% to 95% of mutant alleles (14; 41).
Treatment and management. There is no specific therapy available. Dietary recommendations include reducing fat content (long-chain fatty acids) to less than 20% and increasing complex carbohydrates to approximately 70% (100). Other measures include avoidance of strenuous exercise and fasting. When engaging in exercise, carbohydrates are especially important prior to and during prolonged activity (42).
An anaplerotic diet via supplementation with triheptanoin (medium-chain triglyceride) modestly improved exercise tolerance in carnitine palmitoyltransferase II deficiency and other long-chain fatty acid oxidation disorders (86; 30).
Experimentally, fibrates can increase fatty acid oxidation, and initial reports showed a benefit in open-label studies (11). However, a randomized double-blind clinical trial with bezafibrate did not demonstrate clinical efficacy in carnitine palmitoyltransferase II deficiency and other fatty acid oxidation disorders (75). Resveratrol, a compound that enhanced fatty acid oxidation in animal studies, did not improve exercise capacity in a randomized, double-blind, and cross-over clinical trial (101).
Clinical presentation. Most cases present in the neonatal period with frequent hypoketotic hypoglycemia, respiratory distress, hypothermia, progressive cardiomyopathy, liver failure, and a high mortality rate (59). A rare, less severe form has been reported, with onset in early childhood characterized by episodic hypoketotic hypoglycemia with hyperammonemia and rhabdomyolysis triggered by fasting or febrile illness (80). It can resemble carnitine palmitoyltransferase II deficiency, but the phenotype is typically more severe.
Etiology and pathogenesis. It is caused by a homozygous defect in the SLC25A20 gene. The translocase deficiency results in long-chain fatty acids that do not reach the mitochondrial matrix for beta-oxidation and consequent energy failure.
Evaluation and diagnosis. There is a secondary reduction in serum carnitine levels, and the acylcarnitine profile demonstrates elevated levels of long-chain species (C16 and C181), with a pattern similar to carnitine palmitoyltransferase II deficiency. Urine organic acids can reveal nonspecific dicarboxylic aciduria. DNA testing or enzymatic studies on skin fibroblasts are required for confirmation.
Treatment and management. Carnitine and medium-chain triglyceride supplementation and a low-fat, high-carbohydrate diet are the mainstays of treatment (80).
Clinical presentation. Three main clinical phenotypes have been described: (1) a severe neonatal form with recurrent Reye-like encephalopathy and cardiomyopathy associated with a high mortality rate; (2) an intermediate early-onset form with recurrent rhabdomyolysis with hypoketotic hypoglycemia and respiratory failure; and (3) a milder late-onset form with myoneuropathic symptoms (99; 72).
The first symptoms of the mild late-onset form appear between 1 and 6 years of age, which is earlier than the myopathic form of other fatty acid oxidation defects (10). Peripheral neuropathy usually precedes the development of rhabdomyolysis (99). Sensorimotor peripheral neuropathy is the most common pattern, but pure sensory or motor neuropathy variants can be seen as well (34). Recurrent rhabdomyolysis is frequently triggered by exercise, febrile illness, cold exposure, or prolonged fasting; it is associated with respiratory failure in half of the cases (72). An unusual clinical subtype involves recurrent periodic paralysis-like illness, rather than rhabdomyolysis, triggered by febrile state. This could be explained by the thermosensitive characteristic of trifunctional protein (02).
Progressive sensory-motor axonal neuropathy is a common clinical feature that can help distinguish mitochondrial trifunctional protein from other fatty acid oxidation defects (10). Secondary Achilles tendon contractures can develop in some patients (99). Pigmentary retinopathy due to long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency has frequently been observed in moderate or severe disease (29). Hypoparathyroidism seems to be more frequent in Japanese patients compared to Caucasian reports (10).
Etiology and pathogenesis. Mitochondrial trifunctional protein is a hetero-octamer located in the inner mitochondrial membrane and encoded by two nuclear genes. Its enzymatic function, downstream from very long-chain acyl-CoA dehydrogenase, catalyzes the last three steps of long-chain fatty acid beta-oxidation: long-chain enoyl-CoA hydratase, long-chain 3-hydroxyacyl-CoA dehydrogenase, and long-chain 3-ketoacyl-CoA thiolase. This disease can be classified into two groups: (1) isolated long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency due to alpha subunit mutation (HADHA) and (2) combined or complete mitochondrial trifunctional protein deficiency due to alpha or beta subunit mutations (HADHA or HADHB).
Evaluation and diagnosis. As with other fatty acid oxidation disorders, acylcarnitine profile by tandem mass spectrometry is important for diagnosis, and the yield is increased if performed during fasting or metabolic crisis. There is an elevation of long-chain plasma 3-hydroxy-acylcarnitines (3OH-C16, 3OH-C18) and 3-hydroxydicarboxylic aciduria. Total serum carnitine is usually reduced.
Given the superimposed peripheral neuropathy, muscle biopsy may show a myopathic pattern mixed with neurogenic changes, and MRI T1W/STIR sequences typically show signal change in both proximal and distal muscles (19). Lipid accumulation is rarely seen, but type 1 fiber predominance is common (99). Of note, there is a reduction of activity in respiratory chain complexes (RCCI-III) and ragged red fibers in long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency (16).
Diagnostic confirmation can be achieved by enzyme assay on skin fibroblasts or blood genetic testing (99). Genetic analysis on muscle tissue is not required because the mutation is in nuclear DNA and is not isolated to myocytes. The most common mutation in isolated long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency is c.1528G>C; p.Glu510Gln, which is present in approximately 90% of mutant alleles of Caucasian patients (72). Mutations in combined mitochondrial trifunctional protein deficiency are more heterogeneous, both in Caucasian and Japanese reports (12; 10).
Treatment and management. There is no specific treatment. A high-carbohydrate and low long-chain fat diet and avoidance of fasting or other metabolic stressors are the mainstays of treatment. These measures can reduce the frequency of rhabdomyolysis and of the potentially fatal respiratory failure associated with metabolic crisis (99). Diet modification does not seem to reduce retinopathy or peripheral neuropathy risk (100).
Triheptanoin and docosahexaenoic acid supplementation have been proposed for mitochondrial trifunctional protein and long-chain 3-hydroxyacyl-CoA dehydrogenase deficiencies in children (106; 100).
Clinical presentation. LPIN1 recessive mutations are responsible for more than 50% of recurrent childhood rhabdomyolysis cases in Western countries, but the incidence in Asia is low (63; 13). Patients with homozygous mutations present in early childhood (2-7 years of age) with recurrent rhabdomyolysis that is most commonly precipitated by febrile illness (124). Other triggering factors include fasting, prolonged exercise, or general anesthesia. Rhabdomyolysis is usually severe (> 10K IU/L) or massive (CK> 100K IU/L), with a mortality rate of up to 30% (63; 64). Between episodes, the neurologic exam is normal, and creatine kinase levels are normal or slightly elevated (< 700 IU/L). A mild, chronic, fixed myopathy can develop in patients entering the fourth or fifth decades of life (63). Symptoms are confined to skeletal muscles, except for rare cardiomyopathy (63). In 40% of cases, heterozygous patients for LPIN1 can develop myalgias, cramps, and mild muscular symptoms, or they may be susceptible to statin-induced myopathy (124; 63).
Etiology and pathogenesis. The LPIN1 gene encodes for muscle-specific phosphatidic acid phosphatase, a key enzyme in triglyceride and membrane phospholipid synthesis. Specifically, it catalyzes the conversion of phosphatidic acid to diacylglycerol in the cytosolic triacylglycerol synthesis pathway. Diacylglycerol is the precursor of essential plasma membrane phospholipids, including phosphatidylcholine and phosphatidyl-ethanolamine. A secondary accumulation of lysophospholipids could also contribute to cell damage as they can act as detergents or perturb signal transduction (124). Besides the cytosolic enzymatic function, LPIN1 in the nucleus works as a transcriptional coactivator of genes involved in energy pathways (102).
The LPIN2 isoenzyme is expressed in adipose tissue, and its preserved function likely accounts for the lack of serum lipid abnormalities or lipodystrophy in the LPIN1 phenotype (64). LPIN2 deficiency leads to chronic recurrent multifocal osteomyelitis and Majeed syndrome (congenital dyserythropoietic anemia).
Evaluation and diagnosis. Serum carnitine levels, plasma acylcarnitine profile, plasma amino acids, and urinary organic acids are normal. Muscle biopsy typically reveals increased lipid content in 75% of patients (mild to moderate lipoidosis), type 1 muscle fiber predominance, and moderate atrophy of type II fibers (63). A small percentage shows ragged-red fibers or occasional COX-negative fibers.
Diagnosis is confirmed by the demonstration of homozygous or compound heterozygous mutations in the LPIN1 gene. An intragenic deletion (c.2295-863_2410-27del) is the most common cause in Caucasians (63).
Treatment and management. There is no specific treatment for LPIN1 deficiency. High carbohydrate intake during catabolic states (exercise, fasting, or febrile illness) and intravenous glucose infusion during rhabdomyolysis have been advocated in a case series (79). Aggressive hydration and close monitoring for hyperkalemia (and arrhythmias) are important during febrile illness-induced rhabdomyolysis related to LPIN1 deficiency as these episodes have a high mortality (63). Corticosteroid treatment during the acute period of rhabdomyolysis was associated with greater survival in a retrospective study (110), but findings need to be confirmed in a prospective trial.
Differential diagnosis. Recurrent rhabdomyolysis is a frequent feature of metabolic myopathies. The differential diagnosis includes defects in lipid metabolism, glycogen breakdown, mitochondrial fatty acid oxidation, and oxidative phosphorylation.
Among the lipid metabolism disorders, milder late-onset forms of long-chain fatty acid oxidation defect, including very long-chain acyl-CoA dehydrogenase deficiency, carnitine palmitoyltransferase II deficiency, mitochondrial trifunctional protein deficiency, and LPIN1 deficiency should be considered. LPIN1 deficiency is a major cause of recurrent severe or massive early childhood rhabdomyolysis triggered by febrile illness in Western countries.
Carnitine palmitoyltransferase II deficiency and McArdle disease are the most common entities among lipid and glycogen disorders, respectively. In patients with myophosphorylase deficiency, myoglobinuria is invariably triggered by exercise, which is usually of high intensity. However, patients with carnitine palmitoyltransferase II deficiency have episodes of myoglobinuria not only after exercise (which can be of moderate intensity but is usually prolonged), but also after prolonged fasting or febrile illness without exertion, or after a combination of factors. The forearm exercise test (grip test) is a common investigation performed in the work-up of patients presenting with exercise intolerance and rhabdomyolysis. It consists of the measurement of lactate and ammonia from the antecubital vein before and after exercise. Normally, a nearly 4-fold increase in lactate and a 2- to 3-fold increase in ammonia are present after exercise. The forearm exercise test shows a normal rise in patients with carnitine palmitoyltransferase II deficiency, but there is typically a null lactate response (< 2.0 fold) with a normal or excessive ammonia peak (> 3 fold) in McArdle disease. Unlike McArdle disease, the creatine kinase level is typically normal between attacks in carnitine palmitoyltransferase II deficiency. During rhabdomyolysis, an abnormal acylcarnitine profile is seen in carnitine palmitoyltransferase deficiency but not in McArdle disease.
Mitochondrial trifunctional protein deficiency causes a unique phenotype, which closely resembles hereditary sensory-motor neuropathies (ie, Charcot-Marie-Tooth) or spinal muscular atrophy.
Mitochondrial disorders typically cause static symptoms with isolated muscle weakness or a multisystem disorder. However, cytochrome B (complex I) deficiency due to a mutation in the mitochondrial genome can present with episodic rhabdomyolysis and exercise intolerance, mimicking fatty acid oxidation disorders. MELAS features include exercise intolerance among typical CNS symptoms (stroke-like episodes, epilepsy). There is increased creatine kinase, elevated serum fasting lactic acid with a high lactic and pyruvic acid ratio, and an abnormal urinary organic acid profile that can point to the diagnosis, which is established by muscle biopsy and genetic testing.
Muscular dystrophies can, on occasion, cause recurrent rhabdomyolysis. Recessive limb-girdle muscular dystrophy (LGMD2B or dysferlinopathy), dystrophinopathies, or RYR1 mutations are the most noteworthy. Typically, there is some degree of baseline muscle weakness and an elevation of creatine kinase levels that points to the diagnosis.
Congenital myotonia is characterized by pronounced muscle stiffness that can somewhat resemble muscle pain and contractures present in metabolic myopathies. However, stiffness from myotonia typically improves with exercise (“warm-up” phenomenon) as opposed to muscle pain that worsens with exercise in lipid myopathies. EMG can be helpful in distinguishing these conditions as diffuse myotonic discharges are present in Becker or Tomsen types of congenital myotonias.
When the clinical presentation (exercise intolerance, recurrent rhabdomyolysis, hypoketotic hypoglycemia) is suggestive of a fatty acid oxidation disorder, plasma acylcarnitine analysis is the best initial step in diagnosis. Plasma is preferred to dried blood spot as it provides a higher sensitivity. In general, the diagnostic yield is increased if the analysis is performed during fasting or an acute event. Acylcarnitine profile is also used for newborn screening in some countries, including the United States.
Urinary organic acid analysis is also helpful in conditions that cause exercise intolerance and episodic rhabdomyolysis with dicarboxylic aciduria (multiple acyl-CoA dehydrogenase deficiency, mitochondrial trifunctional protein). The diagnostic approach can be determined based on clinical phenotype, acylcarnitine profile, and urinary organic acid profile. A normal muscle biopsy or the lack of increased lipid content does not rule out a fatty acid oxidation defect. Genetic testing is the preferred method of diagnostic confirmation due to its convenience and precision. A suspected entity locus can be tested separately or as part of next-generation targeted sequencing panels that allow simultaneous analysis of multiple genes known to cause recurrent rhabdomyolysis or exercise intolerance (97).
Functional enzymatic assessment of beta-oxidation components in muscle, lymphocytes, or skin fibroblasts can provide diagnostic confirmation but is usually only available at reference laboratories. Therefore, this is usually reserved for cases in which genetic testing is negative or inconclusive, or when a new mutation is suspected.
Bortezomib is a proteasome inhibitor and antineoplastic agent used for the treatment of refractory multiple myeloma and certain lymphomas. The peripheral neurotoxicity of bortezomib is very well documented. Distal painful sensory polyneuropathy has been associated with this medication in a dose-dependent manner (55).
In a cohort of multiple myeloma patients treated with bortezomib, 50% developed a clinically significant myopathy (35) manifested by proximal leg weakness with normal creatine kinase that developed early in the course (within the first two cycles). Muscle biopsy performed in a patient revealed lipid accumulation and ultrastructural mitochondrial changes. The myopathy was reversible with discontinuation of the drug. The exact mechanism of this type of toxicity is unclear, and the high frequency of myopathy found in this cohort has not been replicated.
Zidovudine was the first antiviral approved for the treatment of HIV. This drug is no longer a first-line agent but is still used in combination with newer drugs. The primary myopathy associated with this drug has features of mitochondrial dysfunction with “ragged-red” fibers. Other features include an increase in muscle lipid content and a reduction in muscle carnitine levels, which are possibly due to a reduction in mitochondrial long-chain fatty acid oxidation (15).
Primary carnitine deficiency may be uncovered or can worsen during pregnancy due to a normal carnitine reduction in this physiologic state. The primary myopathic form of coenzyme Q10 may also be unmasked during pregnancy (37; 27). For at-risk pregnancies, antenatal diagnosis can be pursued through DNA testing from amniocenteses or villus sampling.
In a study, pregnancies in mothers of very long-chain acyl-CoA dehydrogenase deficiency neonates showed no complications (89). Delayed myalgias or rhabdomyolysis after the effort of delivery have been reported in patients with very long-chain acyl-CoA dehydrogenase deficiency (47). Preeclampsia; hemolysis, elevated liver enzymes, low platelet count (HELLP) syndrome; and preterm labor have been reported in mothers carrying infants with long-chain acyl-CoA dehydrogenase or mitochondrial trifunctional protein deficiencies (99; 68; 10).
General anesthesia and the prolonged fasting required prior to surgery can trigger episodes of rhabdomyolysis and secondary renal failure in long-chain fatty acid disorders (45). However, these patients do not seem to be at risk for malignant hyperthermia (122).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.
Ezequiel A Piccione MD
Dr. Piccione of University of Nebraska has no relevant financial relationships to disclose.
See Profile
Aravindhan Veerapandiyan MD
Dr. Veerapandiyan of University of Arkansas for Medical Sciences has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neuromuscular Disorders
Jun. 16, 2026
Neurogenetic Disorders
Jun. 01, 2026
Neuromuscular Disorders
May. 27, 2026
Neuromuscular Disorders
May. 21, 2026
Developmental Malformations
May. 08, 2026
Neurogenetic Disorders
May. 08, 2026
Neurogenetic Disorders
Apr. 30, 2026
Neuropharmacology & Neurotherapeutics
Apr. 23, 2026