



Neurobehavioral & Cognitive Disorders
Mental status examination
Jun. 17, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
In this article, the author describes olfactory bulb agenesis and dysgenesis and also reviews the embryology, maturation, neuropathology, and clinical diagnosis and manifestations. The olfactory bulb is still immature but functional in the term neonate. It is a reservoir of progenitor “stem” cells capable of neuronal differentiation in the adult as well as in the fetus. Complete agenesis of the olfactory tract results in congenital anosmia. Olfactory reflexes in the neonate and preterm infant can be tested as early as 28 weeks’ gestation using a nonirritant aromatic substance, such as peppermint (infantile olfactory reflexes). Fetuses perceive odors in the amniotic fluid that circulate through the nasal passages in the late second and third trimesters. Agenesis of the olfactory bulbs (arrhinencephaly) is a constant feature in alobar and semilobar holoprosencephaly but also can occur as an isolated minor cerebral malformation without other dysgeneses. At times it is asymmetrical. In septo-optic-pituitary dysplasia the olfactory bulbs often are hypoplastic. In Kallmann syndrome, there is hypogonadism with delayed or absent puberty; associated neurologic findings may be present, such as ataxia, impaired hearing, eye movement disorders, or mental handicap. Diagnosis is confirmed by MRI, and molecular studies can reveal genetic mutations. Dysgenesis, rather than agenesis, of the histological architecture of the olfactory bulbs and synaptic circuitry occurs in some genetic syndromes, such as fragile X, and particularly in postzygotic somatic mutations including tuberous sclerosis complex, hemimegalencephaly, and focal cortical dysplasia type II. Multiple genetic factors are essential in normal olfactory bulb ontogenesis. Supernumerary olfactory bulbs are rarer than agenesis but documented and associated with impaired olfaction. Fusion of olfactory bulbs may occur in some cases in which the bulb appears unilaterally absent on gross examination.
|
• Agenesis of the olfactory bulbs (arrhinencephaly) may occur as an isolated defect, as a component of holoprosencephaly (regardless of the genetic etiology) associated with other cerebral malformations, or as a component of Kallmann syndrome with endocrinopathies. | |
|
• Agenesis or hypoplasia of one or both olfactory bulbs at times occurs in isolation without other CNS anomalies or with other focal defects such as hypoplasia of the ipsilateral optic nerve and in septo-optic-pituitary dysplasia. | |
|
• The clinical feature is anosmia, but patients rarely complain of this because it is congenital rather than acquired after they had experienced a sense of smell; the deficit is nonprogressive. | |
|
• It can be diagnosed by MRI of the olfactory bulbs following clinical demonstration of absent olfaction bilaterally; neuropathological confirmation is demonstrated at autopsy, though olfactory agenesis is not a cause of death. | |
|
• Olfactory reflexes are present at birth (after 32 weeks gestation) and can be tested as part of the neurologic exam in neonates and young infants by using an aromatic, non-irritative test substance that does not stimulate the pain endings of the trigeminal nerve in the nasal mucosa; responses are sucking or arousal withdrawal. | |
|
• Olfactory thresholds are determined in part by specific receptors of primary olfactory neurons in the nasal mucosa. | |
|
• The principal well-documented olfactory dysplasias are the presence of a deep longitudinal sulcus on the inferior surface with synaptic glomeruli lining the neural tissue on either side (hemimegalencephaly), megalocytic and dysplastic neurons (tuberous sclerosis and hemimegalencephaly), fusion of the two olfactory bulbs, and delayed maturation of expression of neuronal proteins and synapse formation. | |
|
• Olfactory bulb agenesis should be distinguished from acquired causes of anosmia, such as fracture of the cribriform plate in head trauma, tumors, granulomas and other lesions of the olfactory bulbs or tracts, olfactory groove meningioma, vascular anomalies associated with a persistent fetal olfactory artery, and olfactory impairment due to chemotherapy or other drugs. | |
|
• Multiple genetic mutations and deficiencies of transcription factors and cell adhesion molecules are associated with olfactory agenesis, but usually with other CNS anomalies. | |
|
• Supernumerary olfactory bulbs occur rarely. | |
|
• Some olfactory auras in focal epilepsy may originate in the olfactory bulb and can only be mediated by the amygdala, insula, or entorhinal cortex. | |
|
• Impaired olfaction and olfactory nerve and bulb agenesis may accompany nasal and facial clefts, choanal atresia, and other nasofacial anomalies. | |
|
• Olfaction often is impaired in some infections such as COVID-19, influenza, and cytomegalovirus. |
Perception of odorous soluble molecules is the earliest special sense to develop both phylogenetically and ontogenetically. Even simple medusae (jellyfish) and polyps (hydra) that poses only a simple nerve net without a brain or even a ganglion can perceive and distinguish molecules in surrounding sea water as attractants (ie, food) or adversive elements (ie, potentially harmful) and react accordingly, as noted as early as 1849 by the famous English biologist of his era, Thomas Huxley (96). Human fetuses also perceive odors dissolved in amniotic fluid. Whether these primitive perceptions are best classified as olfactory or gustatory (taste) is a difficult distinction. Both fetuses and preterm neonates distinguish different types of both pleasant and unpleasant odors (129; 200; 214; 144; 213).
The cranial nerves were numbered I to XII by Sömmerring in 1791, but the human olfactory nerve had been described half a century earlier by Winslow in 1733. In ancient times Alcmaeon of Croton (c490-430 B.C.), the father of Greek medicine, first postulated that “channels” (ie, passages, ducts) connected sense organs of the head (nose, eyes, ears, tongue) with the brain, and he is attributed to have first described the olfactory and optic nerves, as reported by Aristotle’s successor Theophrastus (c327-287 B.C.) and later also acknowledged by Galen in the 2nd century A.D. (232).
The first report of anosmia associated with hypogonadism was in 1856 (138). In 1918 Blakeslee described specific anosmia for either pale pink or red verbenas, but not for both, in a group of healthy people (31), and a few reports have since appeared describing familial congenital anosmia in otherwise mainly asymptomatic people (140; 135; 224). In addition, there have been occasional descriptions of "sporadic cases of Kallmann syndrome," in which isolated congenital anosmia is also present (89). In a seminal paper on the genetic aspects of primary eunuchoidism, Fritz Kallmann and colleagues described 36 males and 12 females in whom the condition was associated with combinations of anosmia, color blindness, synkinesis, and mental handicap (102). Subsequent clinical descriptions have permitted better delineation of the syndrome and its association with aplasia or hypoplasia of the olfactory bulbs and tracts (226; 142; 254; 89). A study that investigated isolated congenital anosmia without chromosomal disorders (02) compared the MRI appearances of the frontobasal structures of 16 patients with controls. In half the patients there was bilateral hypoplasia of the olfactory bulbs, and in the remainder there were combinations of hypoplasia and aplasia of the bulbs. Descriptions of anosmia and olfactory aplasia as one component of the holoprosencephalies represent the other extreme of the spectrum (78; 131). Isolation of the Kallmann syndrome gene in X-linked Kallmann syndrome has expanded our insight into this syndrome of anosmia and hypogonadism (133; 177). Septo-optic-pituitary dysplasia is a frequent cerebral malformation and is often accompanied by olfactory bulb hypoplasia or even aplasia (209; 25).
Olfaction and taste are functionally closely related despite the wide separation of their neuroanatomical receptive centers in the olfactory bulb and lower brainstem, respectively (203). Even the simplest marine animals with only a primitive nerve network and no central nervous system, such as medusa (jellyfish) and polyps (hydra, sea anemones, etc.), perceive molecules dissolved in the water as either threatening, resulting in retraction of tentacles, or food, resulting in feeding behaviors. In humans, postnatal respiration is essential for smell, and retronasal olfaction is dependent on expiration and also refines perception of taste for chocolate and other flavors, even accompanied by EEG changes (146). Olfaction is an important aspect of wine tasting and distinguishing foods.
There are no published consensus criteria of the definition of a “cranial nerve.” The following is a suggested working definition for sensory cranial nerves: (a) peripheral nerves; (b) arising from neurons in the peripheral nervous system that possess dendritic receptors for specific sensory stimuli; (c) include a ganglion at some point along the nerve (eg, trigeminal gasserian, semilunar ganglion, facial geniculate ganglion, vestibular/cochlear spiral ganglion, glossopharyngeal jugular or Müller ganglion, vagal jugular, nodose ganglion); and (d) enter the brain, not the spinal cord (207). By these simple criteria, the olfactory is a true cranial nerve, even though its dispersed axons do not become compacted until they reach the olfactory bulb to form layer one of this structure. Though the olfactory nerve lacks a compact ganglion along its course, unique synaptic glomeruli on the ventral surface of the olfactory bulb, facing the cribriform plate of the ethmoid bone through which individual olfactory nerve fibres pass, are connections between primary olfactory axons and dendrites of mitral and tufted cells (203); these glomeruli serve the same function, hence, an olfactory ganglionic equivalent is incorporated into the olfactory bulb as layer two of the bulb (207). Ironically, the accessory olfactory bulb, a transitory fetal structure in humans, has peripheral ganglia like those of other sensory cranial nerves but lacks synaptic glomeruli because it is fused to the principal olfactory bulb far from the cribriform plate because the accessory bulb is fused to the principal olfactory bulb dorso-medially far from the cribriform plate (187; 188). Many residents and attending pediatric neurologists hold the premise that the olfactory nerve is not a “true” cranial nerve, but objective neuroanatomical criteria dispel this unjustified notion in comparing the olfactory with lower sensory cranial nerves. The olfactory bulb and tract are still immature at birth with incomplete synaptogenesis and no myelination, yet already are functional to perceive olfaction from about 28 weeks’ gestation.
Olfactory reflexes in the neonate are reliable and reproducible, demonstrating olfactory perception and discrimination even in premature patients of 30 weeks’ gestation or more (200); furthermore, olfactory responses are elicited in fetuses in utero after 30 weeks, provoked by odors dissolved in the amniotic fluid from certain foods the mother has ingested such as garlic, onion, and curries (206). The observation of olfactory responses in young infants has been confirmed by other authors (223).
Apart from genetic mutations and ischemic infarction from congenital infections, embryonic or fetal exposure to certain teratogens or toxins (for example, arsenic) can interfere with olfactory neuronal differentiation, synapse formation, and result in impaired olfactory perception even into adult life (233).
In adult neurodegenerative diseases such as Alzheimer and Parkinson diseases, impairment of olfaction is not only frequent but one of the first symptoms to appear. Lewy-type a-synuclein pathology in the olfactory bulb, olfactory mucosa, and olfactory areas of cortex (entorhinal; orbitofrontal) are frequent and appear early (132; 147; 36). Activation of dopamine D2 receptors can counteract some olfactory dysfunction in human Parkinson disease (150). Not only are neurons affected but astrocytes also show proteomic alterations in experimental murine models (185).
Despite the ease and reliability of testing in infants and children, regrettably cranial nerve I is the least clinically tested cranial nerve in neonates and at older ages by clinicians, even in cases of cerebral malformations or genetic/metabolic diseases in which its smell is likely to be impaired or absent (208). Similarly, radiologists inconstantly describe the olfactory bulbs and tracts in MRI reports, and neuropathologists usually mention their macroscopic presence or absence at autopsy but rarely take blocks of this part of the brain for microscopic examination. Thus, neuropathological reports of olfactory dysgenesis are infrequent (203).
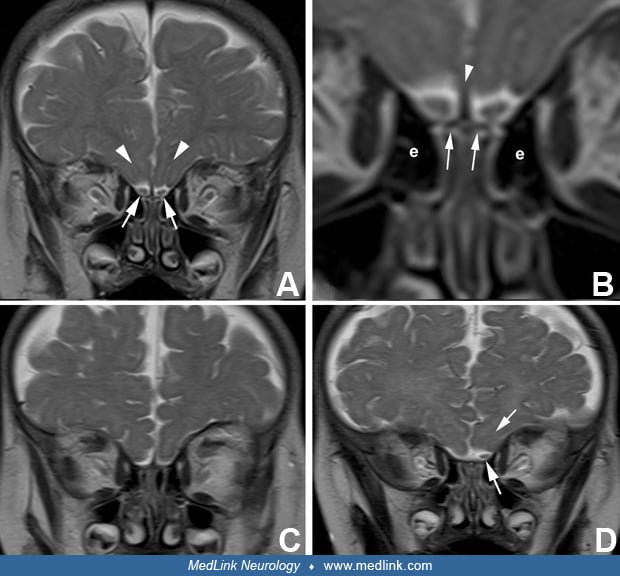
Congenital anosmia as an isolated clinical entity will probably be discovered only by chance during childhood or early adult life and be confirmed by conventional testing (51). Awareness of a positive family history may bring it to light sooner. A limited number of such kindreds have been published (140; 135; 224), and they did not have any relevant associated symptoms. These reports all predated MRI scanning, and there were no reported autopsies, so it is not known whether the olfactory bulbs and tracts were present or absent. Children with apparently isolated congenital anosmia can be demonstrated by MRI to have bilateral aplasia of the olfactory bulbs (15; 198). Unilateral olfactory bulb hypoplasia also can be shown.
Functional MRI studies of congenital hyposmia compared with normal subjects and others with acquired hyposmia have shown that brain activation was present in response to odors in congenital hyposmia; although lower than normal, the distribution was similar to the other (87). Some anosmia is specific or partial; otherwise, normal subjects are unable to perceive certain smells, with musk, sweat, hydrogen cyanide, trimethylamine, and isovaleric acid being the more common. A survey carried out by the National Geographic magazine on 1.5 million people in 1986 found that 35% could not smell sweat, 29% could not perceive musk, and 13% were unable to smell either (81; 82). More commonly, olfactory aplasia with anosmia is part of Kallmann syndrome and associated with a deficiency of gonadotropic-releasing hormone due to a failure of migration of GnRH precursors that should be initiated by intact olfactory axons (52). Kallmann syndrome is one manifestation of the arrhinencephaly spectrum; therefore, more widespread evidence of migration failures such as the holoprosencephalies and other developmental anomalies may sometimes be present (62; 131).
An intrinsic vasopressin system in the olfactory bulb also is demonstrated as a peptide that may be defective in olfactory bulb agenesis; vasopressin appears to be involved in social recognition in addition to its functions in autonomic control of blood vessels (237). Regarding olfaction and cognitive functions, patients with schizophrenia have a higher than expected incidence of olfactory defects, both structural (such as olfactory bulb hypoplasia or agenesis) and strictly functional (241; 196). Olfactory bulb dysgenesis also can be associated with infantile autism and autonomic dysregulation (37).
A contribution from the trigeminal system in some confirmed cases of Kallmann syndrome indicates that there can be a certain ability to distinguish some odorants such as menthol, eucalyptus and alcohol (121). Clinical suspicion of Kallmann syndrome should be present in any case of delayed puberty (particularly in males), although the extent of the delay can vary from extremes of absent or minimal pubertal development to normal gonadal function. This should lead to standard testing of the sense of smell. If this is absent, a review for any possible associated anomalies should be undertaken. Olfactory bulb hypoplasias or agenesis, associated with hyposmia, is frequent in hypogonadotropic hypogonadism (60). Neurologic associations in some, but not all, cases include congenital midline cerebral and cerebellar defects, especially agenesis of the corpus callosum and vermal hamartomas. Neurologic findings are inconsistent and include cerebellar ataxia (which may be a presenting sign), eye movement abnormalities including nystagmus, spasticity, synkinesis, and mental retardation (218; 195). The X-linked form may be associated with renal abnormalities; another variant of Kallmann syndrome associated with X-linked ichthyosis, mental retardation, and short stature has been described (230). Other reported findings include color blindness (102), sensorineural hearing loss (254), cleft lip and palate (226), insulin-dependent diabetes mellitus, and congenital heart disease (50). Associations with anorexia nervosa, cardiomyopathy, and ulcerative colitis have also been reported.
Patients with schizophrenia have impaired olfactory perception and olfactory habituation, particularly in patients with olfactory hallucinations (227). The mechanism may be related to abnormal proteoglycans in the olfactory epithelium (167). The principal proteoglycan of brain is keratan sulfate, which provides a template for axonal fascicles and neuroblast migratory pathways, and is normally strongly expressed in primary olfactory nerve axons that form the outer layer of the fetal olfactory bulb and enter olfactory bulb synaptic glomeruli (211). Low olfactory bulb volume also is described in first-degree relatives of patients with schizophrenia (242).
Olfactory perceptive abnormalities also are reported in anxiety and in obsessive-compulsive disorders (48; 103). Haploinsufficiency of the causative Tbr1 gene of autism in mice impairs olfactory discrimination (92). In humans with haploinsufficiency (campomelic dysplasia due to SOX9 mutation), agenesis of the olfactory bulbs often is associated (55).
Supernumerary olfactory bulbs are much rarer than olfactory bulb hypoplasia or agenesis, but the bulbs can be duplicated or even triplicated, associated with abnormal olfactory groove morphology and clinical hyposmia (125). Though the precise genetic defect is not yet known, duplication of structures is associated with upregulation of a gene in the vertical axis of the neural tube and, in the case of the olfactory bulb, a gene following a ventrodorsal gradient of expression (202).
Agenesis of the olfactory bulbs was described in a twin in whom the heart had failed to develop; the brain in this case showed abnormal convolutions and an accelerated development of cortical gyration (257). In general, an association of olfactory bulb hypoplasia or agenesis is found in infants with congenital heart diseases (166).
The primitive olfactory artery originates at the terminal portion of the internal carotid artery in the fetus during the first half of gestation but may persist even into adult life and, rarely, forms an aneurysm associated with an anterior communicating aneurysm that can be successfully treated surgically if diagnosed by MRA or angiography (259).
There are no progressive features to the condition, so in a majority a normal life span can be anticipated. If olfactory agenesis is associated with a multisystemic genetic syndrome such as CHARGE, however, the prognosis may depend on other organ involvement such as congenital cardiac lesions.
A description of a variant form of Kallmann syndrome, in which endogenous gonadotrophin secretion spontaneously returned later in life, suggests that recovery is possible in some (181).
Kallmann syndrome. Kallmann syndrome is a genetic disorder that may be X-linked or inherited as an autosomal disorder, which can be either dominant or recessive. Research suggests that the majority of familial cases of Kallmann syndrome are autosomal in type and that the X-linked form occurs in less than 15% (220; 164). Olfactory dysfunction was reported in 15 of 19 patients with isolated congenital hypogonadotropic hypogonadism, often associated with hypoplastic olfactory bulbs by MRI (117). See the article on Kallmann syndrome for more details.
CHARGE syndrome. CHARGE syndrome (coloboma of the iris, congenital heart disease, choanal atresia, developmental delay, genital hypoplasia, growth restriction, ear malformations and impaired hearing) is associated with anosmia and olfactory bulb hypoplasia and many features of the Kallmann syndrome (172; 10). A murine model of CHARGE syndrome of humans presents with a defect in neural stem cell proliferation and olfaction in the presence of a genetic defect in the chromodomain gene Chd7, and this same mutation may explain some human cases (122). In human CHARGE syndrome, rhinencephalic anomalies are another major criterion of this genetic syndrome and olfaction should be tested in patients in whom this diagnosis is made (43).
Children with idiopathic hypogonadotropic hypogonadism exhibit a broad spectrum of olfactory function with hypo-osmia or anosmia in one third, suggesting a pathophysiological overlap with Kallmann syndrome (126). Holoprosencephaly is a complex malformation generally detected at birth that is associated with absence of the olfactory bulbs and tracts, except in the mildest forms. The etiology is usually genetic, but more than 14 genes are now documented that produce the same or very similar phenotypes and do not predict the severity of the tissue dysmorphology.
Fragile X syndrome is another genetic disease with almost constant impairment of olfaction and also is the most frequent inherited form of cognitive/intellectual deficiency. It is caused by the FMR1 gene that encodes an important RNA-binding protein, FMRP, which regulates expression of multiple synaptic proteins (33). An animal model of fragile-X syndrome, the Fmr1 knockout mouse, exhibits impaired odor discrimination and hyperexcitability of the olfactory bulb (119).
In adult neurodegenerative characterized clinically by dementia, olfaction is nearly always impaired (261).
Ontogenesis. The olfactory bulbs form at 41 days' gestation in the human, shortly after prosencephalic cleavage with the formation of the sagittal interhemispheric fissure that creates paired telencephalic hemispheres. Axons from neurons in the primitive nasal olfactory epithelium extend to the olfactory placode in the rostrocaudal part of the early telencephalic hemisphere before this placode is even differentiated into the olfactory bulb (14). After olfactory bulb development, an olfactory recess extends from the ventrofrontal part of the lateral ventricle into it but is a transitory structure (203). It regresses and leaves clusters of ependymal cells and microcysts in the postnatal period, similar to the central canal of the spinal cord, and these remnants can be demonstrated by neuroimaging and neuropathologically even in the adult olfactory bulb (225). Persistent dilated olfactory recesses can be found not only in humans but also in cats and other mammals (149). An accessory olfactory bulb and its associated vomeronasal organ and nervus terminalis (cranial nerve 0) are an important supplementary olfactory system in many lower vertebrates, believed to be functional during mating, but are only transitory structures in embryonic and early fetal life in humans (204). Odor recognition depends on the binding of an odorant molecule to a human odorant receptor on primary olfactory neurons; such receptors, such as OR5IE2, often are bound to the fatty acid propionate for receptor activation; one of two large families of G-protein-coupled odorant receptors or trace amine receptors that enable olfactory perception (30; 85).
In the fetus, neuroblasts of the olfactory bulb do not originate within the bulb, but stream into it from the periventricular region of the lateral ventricle and especially the lateral ganglionic eminence with secondary radial migration within the olfactory bulb (113; 194; 130). The lateral ganglionic eminence is particularly the source of inhibitory interneurons of the olfactory bulb (35), though some are produced throughout life by intrinsic progenitor cells within the olfactory bulb (250). The gene CHD7 is key in mediating maturation of neuroepithelial cells to olfactory neurons, and its mutation impairs this transition but can be reversed by the inhibition of another gene EZH2 with which it interacts (93).
Though the olfactory bulb was long thought to achieve maturity by 11 weeks’ gestation in terms of histological identification and arrangement of the neuronal architecture (95), studies of immunocytochemical expression of neuronal proteins and transmitters, synaptogenesis, myelination, and persistence of the olfactory ventricular recess at birth demonstrate that the human olfactory bulb is far from mature at term, even if functional in part (106; 203; 203; 206). This immaturity is incomplete synaptogenesis and myelination. In the rat, synaptophysin and synaptoporin (molecular components of synaptic vesicle membranes regardless of the transmitter contained within the vesicles) is similar in late fetal, postnatal, and adult animals in mitral and tufted neurons of the olfactory bulb, but biosynthesis of these glycoproteins by granule and periglomerular neurons occurs mainly postnatally (26). Serotonin (5-hydroxytryptamine) increases synaptic activity of olfactory bulb glomeruli, acting through serotonin receptors on tufted cells and depolarizing some mitral neurons (38). Myelination in the olfactory bulb and tract have not even been initiated at term and occur in the first few postnatal months (203). In addition to neuronal differentiation, migration, and maturation, a diversity of glial cells with different morphologies develop during fetal life and are not fully developed until term (46). In elderly normal humans and mice, there is a natural regressive change with progressively less glial diversity in the olfactory bulb (114).
Despite neuroanatomical and neurophysiological immaturity, the olfactory system is functional to perceive and react to odors not only in the neonate, but also in the fetus in utero after about 28 to 30 weeks’ gestation, as shown in prenatal clinical and real-time ultrasonographic studies (209). Molecules of foods with strong odors that the mother ingests, such as garlic, onion, curries, and spices, are transported trans-placentally and dissolved in the amniotic fluid, which circulates through the nasal passages with fetal respiratory movements. The fetus reacts differentially to different odors, with graded changes in heart rate and movements (129; 88; 151; 214; 213). Preterm and term postnatal infants show a gradient of olfactory responses (136; 200; 144). Whether fetal and neonatal exposures to strong food odors later influence preferences or aversions to certain foods remains speculative. Human olfactory perception is not just an analytical process of molecular detection alone but is influenced by experience and other interpersonal characteristics (29). Diet also may play a role in olfactory dysfunction (228). Nevertheless, odor dynamics also are encoded in the olfactory system and influence related behavior (03).
The olfactory tract is much more than a simple axonal fasciculus to conduct mitral cell axons from the olfactory bulb to the anterior olfactory nucleus, amygdala, hypothalamus, hippocampus, and entorhinal cortex (part of the parahippocampal gyrus). The tract contains as much grey as white matter and includes extensions of the granular cell core of the olfactory bulb at one end and nodules of the anterior olfactory nucleus at the other, in addition to the soma and long processes of progenitor cells (203). The distal part of mitral cell axons as they emerge from the olfactory tract in humans divide into four fascicles, called the medial, lateral, intermediate, and superior olfactory striae, each with a different target structure (57). These striae also can be demonstrated in the living patient by diffusion tensor imaging (70). The olfactory is the only special sensory system to not project to the thalamus for intermediate relay to the neocortex because it contains its own intrinsic thalamic equivalent within the olfactory bulb and tract: (1) the core of GABAergic granular neurons which lack axons and form intrinsic dendrodendritic synapses; (2) periglomerular interneurons that coexpress GABA and dopamine; and (3) the anterior olfactory nucleus which is a series of nodules (203; 206; 207). In the revised scheme of human neural tube segmentation, both the thalamus and olfactory bulb are derived from prosomere 2, hence, embryology also supports the concept of an independent olfactory thalamus (179; 40).
A unique feature of the olfactory nerve is that it appears to be the only sensory cranial nerve that lacks a peripheral ganglion. However, the synaptic glomeruli at the ventral surface of the olfactory bulb serve the same function as a ganglion, but with primitive evolutionary and ontogenetic structure because of a paucity of neural crest in the region of the developing olfactory placode, which has misled some clinicians into questioning whether the olfactory is a “true” cranial nerve (208). Compartmentalization of the olfactory bulb glomeruli occurs in the postnatal period in rats (104; 109). A segmental architecture also occurs in the piriform cortex (entorhinal area), which puts in order afferent fibers from the anterior olfactory nucleus and directs tertiary efferent olfactory axonal projections (217; 173).
The olfactory nerve is also unique in having its axons rather than being grouped as a compact nerve because the axons must pass individually or in small groups through the multiple fenestrations of the bony cribriform plate; however, after entering the cranial cavity they group as a single compact fascicle that forms the first layer of the olfactory bulb (208).
Another unique feature of the olfactory nerve is its regenerative ability, and also, the olfactory bulb is one of only two sites in the adult brain (the other being the hippocampus) in which progenitor cells persist and can proliferate to form new neurons. Hemopexin, a plasma glycoprotein, is required for adult neuronagenesis in the olfactory bulb (267).
The olfactory epithelium also follows a progressive development in fetal life and is not yet fully mature at birth (168; 180; 158; 110). The olfactory epithelium expands greatly in area, enabling a larger number of primary olfactory neurons, with the development of the turbinate bones in the nasal cavity at 8 to 12 weeks gestation; the olfactory epithelium covers most of the superior and part of the middle turbinates, but not the inferior (215). There is continuous turnover of neurons in the olfactory epithelium throughout life with physiological apoptosis and regeneration by progenitor cells (139). Olfactory thresholds are determined in part by specific receptors of primary olfactory neurons in the nasal mucosa (175).
Neuroanatomic studies have shown that the GnRH secreting neurons of the hypothalamus originate in the olfactory placode and migrate across the olfactory bulb to ultimately reach their final location in the hypothalamus. Defective migration of neuroendocrine GnRH cells may contribute to human arrinencephalic malformations (236). In Kallmann syndrome, these cells and nerves apparently develop normally but then fail to migrate, suggesting that the gene encodes a factor needed for the migration of GnRH cells. The beta-secretase enzyme BACE1, deficient in Alzheimer disease, is essential for axonal guidance of olfactory sensory neurons and normal formation of synaptic glomeruli in the olfactory bulb (186). The axonal guidance protein Draxin, related to DSCAM (Down syndrome cell adhesion molecule), also is important in olfactory bulb development as an inhibitory factor that binds multiple netrin receptors; in Draxin knockout mice, neurite outgrowths from olfactory bulb explants are less inhibited than in wild-type mice (07). The transcription factor Tbr2 is expressed in glutamatergic excitatory neurons; mice with deficiency of this factor in mitral and tufted neurons show disruption of the excitatory-inhibitory balance of neural circuitry (153); though it has not been demonstrated in humans, theoretically it could be a genetic cause of impaired olfaction.
Mutations in Kallmann syndrome 1 (KAL 1) gene on the xp22.3 site result in the X-linked form of the syndrome; mutations on the KAL 2 gene (fibroblast growth receptor 1) underlie one autosomal dominant form (66; 212). Abnormalities of both these genes are expressed in a variety of embryonic tissues, thus, leading to the different clinical expressions of the syndrome in addition to the core feature of anosmia. Mutant mice with Pax6 deficiency exhibit decreased volume of the olfactory bulb (but also of the total brain) and altered lamination of the piriform cortex (34). Fibroblast growth factor receptor 1 mutations alone may produce the phenotype of Kallmann syndrome in children (266). The prokineticin receptor PKR2 has been identified in mice with olfactory bulb agenesis and anomalies of the reproductive system (148; 178), and a missense mutation is present in some human cases of Kallmann syndrome, transmitted as an autosomal dominant trait (67; 199). The FGFR1 gene is mutated in some cases of Kallmann syndrome (161).
The detection of Kallmann syndrome mRNA in other tissues including brain, muscle, and kidney suggests that its absence may account for the non-neuronal symptoms found as part of the syndrome (133). Even within the brain, the gene may be important for the development of structures unrelated to the olfactory system or hypothalamus, such as the inhibitory fibers of the corpus callosum that suppress synkinetic or mirror movements (54). Dental agenesis occurs in some individuals with FGFR1 mutations (21).
Not all cases of olfactory bulb agenesis are Kallmann syndrome. This minor malformation also can occur as an isolated defect in brain development and, with the exception of anosmia, is not associated with other brain malformations or with clinical neurologic deficits, including epilepsy, developmental delay, mental retardation, or cognitive and memory problems (64; 41). The diagnosis is confirmed by clinical demonstration of anosmia (testing of cranial nerve 1) and by MRI with special attention to the olfactory bulbs. Neuropathologically, it can be demonstrated in fetuses, infants, and children not only by absence of the olfactory bulbs and tracts but also by absence of the olfactory grooves at the base of the frontal lobes where these structures normally lie. Olfactory agenesis is accompanied microscopically by hypoplasia, neuronal deficiency, and disorganized laminar architecture of the entorhinal cortex (parahippocampal gyrus), a principal projection site of olfactory tract axons.
Some other genetic mutations in which agenesis of the olfactory bulbs are an associated anomaly are Prader-Willi syndrome (108), Waardenburg syndrome associated with SOX10 mutation (71), and Bardet-Biedl syndrome (231). Mendelian transmission traits that also involve epigenetic mechanisms occur in several chromosomopathies: Rubinstein-Taybi, CHARGE, Kleefstra, and Weaver syndromes (10).
Olfactory receptor neurons project from the sensory epithelium to stereotypical targets within the olfactory bulb. Studies have demonstrated, unexpectedly, that there is a somatotopic spatial map – with a precision similar to that found in the visual and auditory systems – that enables the distinction of thousands of odors (128; 61). Single neuron representations of odors in human piriform cortex and medial temporal lobe are also demonstrated to determine electrophysiological discharge rates (105). The topographic olfactory-epithelium-to-olfactory-bulb map is particularly of interest in terms of plasticity, because there is a constant turnover throughout life of olfactory sensory neurons, both under normal conditions and after injury (16; 58). A protein associated with the fragile-X mental retardation syndrome is one of several that regulate neuronal differentiation in the adult olfactory bulb (219). Nonapoptotic caspase signaling is required for proper neural network formations during olfactory development (162). The HES family of genes regulates sequential stages of neurogenesis in the olfactory epithelium (42). In the ontogenesis of the olfactory bulbs, there is an intimate glial-neuronal interaction in the formation of the synaptic glomeruli (20). Olfactory nerve ensheathing cells differ from the usual Schwann cells of the peripheral nervous system in their interactions with olfactory bulb astrocytes (120).
Entorhinal cortex exerts an inhibitory effect on olfactory input to the basolateral amygdaloid nucleus and piriform cortex (155). Olfactory integration at the cortical level is impaired if the hypothalamic neuropeptide melanin-concentrating hormone is abolished in mice (04). Anosmia and similar cortical olfactory integration occur in a murine model of human fetal alcohol syndrome (255) and after fetal exposure to the neurotoxic effects of lipopolysaccharide in rats (112). Fetal alcohol syndrome also can cause abnormal development of the olfactory bulb with impaired odor discrimination after mice reach maturity (09); the syndrome can also cause defective early olfactory preference learning in early postnatal mice and rats that later can affect behavior (111). Olfactory impairment also can occur in children with autistic spectrum disorder (216).
Another gene recognized in mice, in which poor expression impairs development of the eye and olfactory bulb, is an isoform of Pax6 lacking the paired domain (107). Mice with Pax6 defects show altered murine social interactions in addition to olfactory impairment (53). A neuroligin-3-deficient murine model of olfactory hypoplasia may be a model of a monogenic heritable form of autism (184).
Olfactory bulb agenesis may be associated with agenesis of the corpus callosum and may in part be a disorder of axonal guidance (72). Callosal agenesis and alteration of the anterior commissure, through which some olfactory projections normally pass, may be induced in fetal mice by laser destruction of the primordia of the olfactory bulbs, suggesting that the olfactory bulbs may be one of the induction factors for the forebrain commissures (159). Agenesis of both the olfactory bulbs and corpus callosum similarly is seen in cerebral cortical lissencephaly due to mutation in the TUBA1A gene and is also sometimes associated with ocular anomalies (163; 157) and rhombencephalosynapsis of the cerebellum, with or without holoprosencephaly (98). Agenesis of both the corpus callosum and olfactory bulbs also is reported in an infant with congenital maxillomandibular fusion and duplication of the craniofacial midline (145). Other genetic syndromes with agenesis of hypoplasia of both the corpus callosum and olfactory bulbs are the acrocallosal and Grieg cephalopolysyndactyly syndromes, which involve KIF7 and GLI3 gene mutations in their relation to SHH (229).
Infants with fetal alcohol spectrum disorder may exhibit impaired olfaction (97) but, by contrast, a postnatal enhancing effect on behavioral and neural responses to alcohol, including augmented olfactory perception, is experienced in other patients and is ameliorated by administration of naltrexone (262). Impaired olfaction is common secondary to the cytotoxic effects on both the olfactory nasal epithelium and the olfactory bulbs of chemotherapeutic agents used in the treatment of cancer, with or without anatomical infiltration by neoplastic cells. Olfaction may be altered clinically, with or without olfactory bulb hypoplasia in congenital infections such as cytomegalovirus disease in humans and murine models (123). Haploinsufficiency of a causative gene of autism, Tbr1, results in impairment of olfactory discrimination in mice (92).
Unilateral olfactory bulb agenesis is much rarer than bilateral agenesis but may occur with ipsilateral sinonasal aplasia and orbital cyst and can be diagnosed by high-resolution CT and MRI (247). Asymmetrical olfactory bulb hypoplasia also may occur, at times associated with ipsilateral optic nerve hypoplasia.
The principal well-documented olfactory dysplasias are the presence of a deep longitudinal sulcus on the inferior surface with synaptic glomeruli lining the neural tissue on either side of the sulcus, as in hemimegalencephaly; hamartomatous megalocytic and dysplastic neurons in tuberous sclerosis (59) and in hemimegalencephaly (190; 203); fusion of the two olfactory bulbs; and delayed maturation of expression of neuronal proteins and synapse formation (203). Enlarged olfactory bulbs in hemimegalencephaly may be demonstrated by CT and MRI during life (01) and neuropathologically postmortem (190; 203). Less is known about dysplasias of the olfactory bulb than about other parts of the brain in part because even in autopsies of fetal and infant brains with major malformations, the neuropathological examination of the olfactory bulb is usually limited to a gross description of its presence or absence, and samples for histological and immunocytochemical examination rarely are taken. In tuberous sclerosis complex, another mTOR disorder as a somatic mutation associated with germline mutations of TSC1 or TSC2 genes, olfactory bulb agenesis or hypoplasia is frequent, as demonstrated by MRI (143).
Together with the polymorphic zone in the hilus of the dentate gyrus of the hippocampus, the olfactory bulb is one of the two brain regions in which neuronal regeneration and stem cell utilization is greatest in rodents, nonhuman primates, and humans, after traumatic or hypoxic/ischemic injury (58; 116). This plasticity is important not only for the recovery of olfactory sense, but the olfactory bulb can be an important source of stem cells for transplantation into other parts of the brain and spinal cord after injury. Not only the olfactory bulb, but also the olfactory mucosa is a site of resident stem cells in the mature rodent and human; diseases that affect this sensory mucosa may deplete the population of progenitor cells so that anosmia may result (83). Chemotherapy may abolish or suppress progenitor cells of the olfactory bulb, including in patients in whom the bulb is infiltrated by abnormal cells, as in neurocutaneous melanocytosis (203) or by neoplastic cells in the surrounding leptomeninges.
Olfactory auras in focal epilepsy are well documented after the initial observations by Hughlings Jackson in 1871 (99). In a retrospective study of 806 adult patients with focal epilepsy and an additional 169 with primary generalized epilepsy, the overall prevalence of olfactory auras was 3.9%, and for just those with focal epilepsy, 4.22%, especially if the epileptogenic EEG focus was in the mesial temporal lobe (235). In 1954, Penfield and Jasper, in their classical textbook, noted that olfactory auras could be produced by electrical stimulation of the olfactory bulb during epilepsy surgery (169). These observations and subsequent studies of the development and synaptic organization of the olfactory bulb led to a hypothesis that the olfactory bulb was actually the initial generator of olfactory auras in temporal lobe epilepsy (203) and that the amygdala, insula, or entorhinal cortex mediated the perception and brought it to conscious awareness (206). Impaired olfactory discrimination occurs in adult cases of focal epilepsy, usually arising in epileptogenic foci in mesial temporal lobe structures, including focal cortical focal dysplasias of piriform cortex (at the junction of the temporal and frontal lobes) and hippocampal sclerosis (73). A variety of electrophysiological responses to different odors in adult humans are recorded from the amygdala, hence this structure might be another source of olfactory auras (94). Not only auras but impaired olfactory perception and acuity are experienced by patients with either left or right temporal lobe epilepsy, unrelated to duration and baseline frequency of seizures or type of medication used (63). Olfactory hallucinations were reported in an adult with an anxiety disorder but not epilepsy (100). Basal forebrain electrophysiological recordings from regions regulating olfactory processing demonstrate differences between patients with autistic spectrum disorder and controls (248).
Olfactory hallucinations also are described in patients with migraine, but unlike visual and other sensory auras, they are not presently part of the International Classification of Headache Disorders (49; 08).
In adult neurodegenerative diseases, particularly those associated with alpha-synuclein expression, loss of olfactory sense is a frequent symptom and is diagnostically useful (174). In a murine model of Alzheimer disease, α-synuclean appears early in the olfactory bulbs and extends from there into the cerebrum (244). Certain neurons in the olfactory bulb (external plexiform, granular and glomerular layers) and tract express secretagogin, a cytosolic calcium-binding protein coexpressed with calretinin and beta-III-tubulin, and fail to differentiate terminally for synaptic contact or may lose their connectivity in Alzheimer disease, which may be part of the pathogenesis in neurodegenerative diseases (17). Olfactory discrimination and perception often are diminished or lost in normal aging due to atrophy of both the olfactory mucosa with loss of primary olfactory neurons and also the olfactory bulb (18), and also the loss of diversity of glial cell types (114).
Murine models are available of various human genetic mutations or deletions in which olfaction is impaired and the olfactory bulb is hypoplastic or anatomically altered: fragile-X syndrome (119), Down syndrome (56), KBG syndrome (ankyrin repeat domain 11) (84) and Shank3 deficiency associated with reduction in GABA receptors and in humans with autistic spectrum disorder (152). Olfactory bulb disorders also are associated with facial dysmorphism in chick and pig embryos after embryonic exposure to teratogens (249). Murine models have been valuable in studying the acquired effects of viral infections, including COVID-19, on the structure and metabolism of the olfactory system and effects and limitations of antiviral pharmacological therapies (251).
Kallmann syndrome is rare and occurs in about 1 in 10,000 men and 1 in 50,000 women. The rate of holoprosencephaly and arrhinencephaly in the Atlanta, Georgia, area increased from 0.58 to 1.2 per 10,000 between the years 1968 and 1992 (189). Holoprosencephaly and arrhinencephaly represent 8.5% of total central nervous system malformations in 363 cases, representing 8.8% of 4122 perinatal or neonatal autopsies in a pathological series (170).
Prenatal diagnosis of Kallmann syndrome is possible in the X-linked form by Southern blot analysis of fetal DNA (177).
Rare cases of isolated hypoplasia of the olfactory bulbs may be the result of a local insult to the cribriform plate in early fetal life (131).
In deficiency of GnRH in the male, the testes are normal but remain in the prepubertal state. Absent or delayed puberty should lead to consideration of Kallmann syndrome in either sex as one possible cause, and specific testing for anosmia is confirmatory. Fifty percent of individuals with hypogonadotrophic hypogonadism do not have a positive family history or anosmia; further experience will tell whether these are formes fruste of the syndrome (52). Disorders of the cerebellum or pes cavus occur in 40%, mirror movements in 60% (218), and mental handicap may also be present.
Arrhinencephaly is a constant feature of alobar and semilobar holoprosencephaly, although rudiments of an olfactory bulb may sometimes be recognized histologically near the olfactory trigone region (205). Other midline cerebral malformations, such as septo-optic-pituitary dysplasia, also frequently are accompanied by hypoplasia or aplasia of olfactory structures and the olfactory groove at the base of the frontal lobe (209; 25). Olfactory agenesis is a frequent, but not constant feature of Meckel-Gruber syndrome, which is a complex cerebral malformation that includes encephalocele (06). Dysgenesis, rather than agenesis, of the olfactory bulbs occurs in some genetic syndromes, such as short-rib-polydactyly syndrome, and may be asymmetrical and associated with other cerebral malformations (258). This human syndrome is of interest because there is a murine model of arrhinencephaly and hydrocephalus, also associated with polydactyly (91; 160), and the defect in these mice appears to be downregulation or suppression of the Gli3 gene without alteration of sonic hedgehog (Shh) expression (246; 245). Primary cilia control telencephalic patterning including olfactory bulb formation via Gli3 proteolytic processing (27). The association of polydactyly in both the mouse and human phenotypes suggests that this gene or a related one might also be responsible in humans. A 4-generation family had decreased sense of smell associated with polydactyly and other anomalies (24). The homozygous mouse embryos also develop exencephaly (encephalocele) when exposed to valproic acid (137).
Though impaired olfaction is described in some girls with Rett syndrome, a murine model of Mecp2 genetic mutation exhibited normal mitral cell dendritic development and did not support the hypothesis that this gene plays an important role in olfactory ontogenesis (165).
Olfactory micronodules associated with olfactory impairment may occur in tuberous sclerosis and also in mice with Tsc1 deletions (75).
Bilateral olfactory bulb agenesis was found in seven or eight patients with Waardenburg syndrome, types 2 and 4 associated with SOX10 mutations; they also have impaired hearing and abnormalities of the cochlear nerves and internal auditory canals of the temporal bones (71). SOX10 has the same genetic mutation as in Kallmann syndrome with deafness (171). SOX9 mutations cause campomelic dysplasia and haploinsufficiency with agenesis of the olfactory bulbs (55).
Simple olfactory bulb agenesis without other malformations is rare but known, and might result from a brief single exposure to a teratogen on the 41st day of gestation, or from a limited defect in genetic programming of olfactory bulb development; it also is demonstrated in the brains of some infants with trisomy 13 without other features of holoprosencephaly (201).
Olfactory groove meningioma is the most frequent tumour that could invade the olfactory bulb and cause anosmia, but it would not be a congenital lesion (47). A rarer tumour is olfactory bulb Schwannoma (74). Amongst neurocutaneous syndromes, a dysplasia with neoplastic transformation that infiltrates the olfactory bulbs in early life is neurocutaneous melanocytosis (77). Tumors of the olfactory bulb itself, such as olfactory neuroblastoma, are a rare cause of altered perception or loss of olfactory function (176; 154). Radiotherapy administered to patients with nasopharyngeal neoplasms reduces olfactory bulb volume and diminishes olfactory function (252).
Anatomical anomalies of the nose and face at times are accompanied by olfactory impairment and defects in peripheral olfactory pathways and even the olfactory bulb. Examples are nonsyndromic orofacial clefts (28; 193), deviations of the nasal septum, shallow olfactory fossa and defective cribriform plate (197), and choanal atresia that is part of the CHARGE syndrome (43).
Children with congenital heart disease may exhibit alterations and asymmetry of olfactory bulb volume, which some authors associate with executive dysfunction in adolescents (183). Reduced olfactory bulb volume can also be demonstrated by MRI in chronic viral-induced (COVID-19) olfactory dysfunction (22).
Olfactory reflexes may be tested in the neonate or in infancy using nonirritative aromatic substances. In reality, all molecules that stimulate olfactory receptors also can trigger trigeminal pain receptors in the nasal mucosa, but it is a matter of ratio (68); peppermint is predominantly olfactory, whereas ammonia is predominantly painful. Peppermint elicits sucking or arousal-withdrawal when the child is content, not crying, and not in quiet (deep) sleep. It is a reliable reflex after 32 weeks’ gestation but frequently can be elicited as early as 28 weeks (200). Neonatal olfactory reflexes have been confirmed by other authors (80). This reflex is clinically useful in the assessment of neonates with suspected cerebral malformations, particularly holoprosencephaly because of its association with arrhinencephaly (205).
MRI provides the best objective manner of confirming agenesis or hypoplasia of the olfactory bulbs, even by subjective impression without quantitative volumetry, and they also can assess the olfactory groove for further confirmation (23; 69; 124). The olfactory tract and its striae can be demonstrated by diffusion tensor MRI (70; 22). MRI4 also is useful in children and adults with new onset of olfactory loss of unknown origin (90).
In conventional MRI, the olfactory bulbs are best seen in the coronal plane in T2-weighted images at a level at the posterior half of the orbits that includes the turbinate bones, the horizontally oriented cribriform plate and crista galli, and the vertically oriented plate of the ethmoid bones. The olfactory bulbs also can be identified in the axial plane of MRI. The volume of the olfactory bulb and the depth of the olfactory sulcus can be quantitatively measured (191). A shallow olfactory sulcus is sometimes seen in schizophrenia and may indicate an early embryonic disruption (241). The olfactory recess of the rostral ventral part of the frontal horn of the lateral ventricle is prominent in embryonic life until about 26 weeks’ gestation, but MRI may demonstrate persistence of this olfactory ventricle in some cases postnatally as an incidental asymptomatic finding (225).
Olfactory bulbs may be demonstrated reliably by MRI after 30 weeks gestation, and hypoplasia of these structures also documented by late prenatal and early postnatal MRI (19; 209). On postnatal MRI, normal olfactory bulbs are nearly always identifiable on 3 mm coronal spin-echo T2-weighted images (265). The recognition is easy in infants because of their prominent bifrontal subarachnoid fluid spaces. With 3-dimensional high-resolution T2-weighted images, the olfactory bulbs can be identified in both axial and coronal planes, the latter at the level of the posterior orbits (265). The olfactory tract is distinguished from the bulb by a change in calibre, but a clear neuroanatomical landmark is lacking (32). A normal olfactory bulb is usually accompanied by an olfactory sulcus, previously called the olfactory groove on the ventral surface of the gyrus rectus and in which the olfactory bulb and tract lie, and it is also readily identifiable in both axial and coronal T2-weighted images. Absence of the olfactory bulbs can be bilateral or unilateral, both conditions diagnosed with confidence by MRI. Absence of the olfactory bulb nearly always is accompanied by absence of the olfactory sulcus. Congenital arhinia (absence of the external nose, nasal cavities, olfactory epithelia, and olfactory bulbs) is a rare malformation diagnosed prenatally by fetal MRI (127).
Computed tomography of the anterior cranial base in agenesis of the olfactory bulb reveals hypoplasia of the ethmoid bone, through which the axons of the olfactory receptor cells pass to reach the bulb (141). In congenital anosmia, anterior skull base abnormalities can be demonstrated (260).
Endocrine investigations in the prepubertal age group are unsatisfactory, but in adults the assay of subnormal levels of gonadal steroids and gonadotrophins are straightforward. Screening for possible associated renal malformations is indicated. MRI using coronal slices has demonstrated abnormalities of the olfactory system (115; 239; 264), and other work has shown that there is bilateral absence of the olfactory bulbs, tracts, and sulci in approximately two thirds of the cases of Kallmann syndrome, whereas in the remainder they are only present unilaterally or are hypoplastic or rudimentary (79; 182; 263). In the normal human fetus, olfactory sulci can be reliably detected from 30 weeks’ gestation and the olfactory bulbs from 30 to 34 weeks (19). MRI also is useful for assessing the olfactory bulbs in anosmic patients after traumatic head injury (256). Posttraumatic anosmia may in part be due to damage to cerebral centers of olfaction but not just to the olfactory bulbs (192).
Bilateral responses to unilateral magnetic stimulation of the motor cortex have been reported in the syndrome (54). The X-linked form of Kallmann syndrome has been localized to the Xp22.3 site by genetic linkage studies. Contiguous gene syndromes such as associated steroid-sulfatase deficiency with X-linked ichthyosis, in which there are deletions of neighboring portions of the short arm of the X-chromosome, can be identified by DNA analysis (177).
Vascular anomalies also should be considered in the differential diagnosis of olfactory impairment. The olfactory artery is a primitive transitory fetal vessel arising from the proximal anterior cerebral artery that may persist; MR angiography may demonstrate rare aneurysms of that vessel (259; 243) or even arteriovenous fistulae also involving the anterior ethmoidal arteries (240) or mixed pial-dural arteriovenous malformations that involve a primitive olfactory artery (101).
Congenital infections such as cytomegalovirus may cause olfactory bulb hypoplasia and prenatal infarction. Postnatally acquired cytomegalovirus also can result in olfactory atrophy. COVID-19 infection regularly reduced perception of odor and taste in adults (11; 221; 253), and MRI in anosmic patients with COVID-19 discloses olfactory bulb abnormalities, such as altered signal intensity ratios that are not found in COVID patients with preserved sense of smell (44). In chronic COVID-19, the olfactory bulbs may show reduced volume by MRI (22). Nonolfactory additional MRI findings may include subcortical white matter abnormalities and cerebral microhemorrhages (05). In long COVID-19 and afterward, there often are other symptoms such as chronic headache, mental clouding, and memory and cognition deficits (65; 76; 156). Viral entry into and replication within the sustentacular (ensheathing) cells of the olfactory nerve fibres is the likely mechanism of pathogenesis (222). MRI studies may demonstrate atrophy of the olfactory epithelium and bulbs (45; 234), but these findings may be transitory and recover in follow-up imaging (13). Immunological study of patients with long COVID-19 reveal a likely role of endothelial activation and clotting abnormalities and diverse auto-antibody specificities, but the data remain insufficient for mechanistic synthesis (12).
Clinical testing of olfactory perception and discrimination is useful in adults with suspected dementia or mild cognitive impairment because loss of olfactory is an early sign in senility or neurodegenerative diseases (238).
The delayed puberty in males is managed by replacement therapy, and pulsatile injections of GnRH have been shown to normalize the pituitary-gonadal axis (52). Genetic counseling is indicated. Treatment with zinc is often advocated in elderly patients who develop anosmia and seems effective in some in restoring a sense of smell. However, zinc-containing compounds also may have a cumulative neurotoxic effect on the olfactory neuroepithelium and on mitral cells of the olfactory bulbs in rats, so caution should be exercised in prescribing it for humans (39). In a randomized, placebo-controlled trial of oral zinc supplements for chemotherapy-related anosmia and hypogeusia, no objective benefit was demonstrated (134).
In congenital anosmia not associated with brain malformations or olfactory bulb agenesis, oral theophylline is effective in some cases in restoring smell, probably because it induces olfactory receptor growth (86).
Olfaction cannot be restored in patients with agenesis or severe hypoplasia of the olfactory bulbs, but in some milder cases of olfactory hypoplasia, such as associated with septo-optic-pituitary dysplasia, some sense of smell may develop but lacks the keen olfactory discrimination and low threshold of normal subjects. Acquired damage to the olfactory mucosa, such as in certain viral infections, usually result in recovery of sense of smell, but in a minority of patients may be a permanent olfactory deficit.
The successful induction of ovulation and completion of pregnancy has been reported in a woman with Kallmann syndrome using recombinant human luteinizing hormone and follicle-stimulating hormone (118). Though olfaction has been demonstrated experimentally in human fetuses from 30 weeks’ gestation, this testing is not yet clinically available for application to patient care.
No known increased anesthetic risk is known in simple arrhinencephaly, but other associated malformations of the brain and other organs may cause an increased risk.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Harvey B Sarnat MD FRCPC MS
Dr. Sarnat of the University of Calgary has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neurobehavioral & Cognitive Disorders
Jun. 17, 2026
Neuro-Oncology
May. 27, 2026
Neuropharmacology & Neurotherapeutics
May. 14, 2026
Developmental Malformations
May. 08, 2026
Developmental Malformations
May. 08, 2026
Developmental Malformations
May. 08, 2026
Neuro-Oncology
Apr. 30, 2026
Developmental Malformations
Apr. 24, 2026