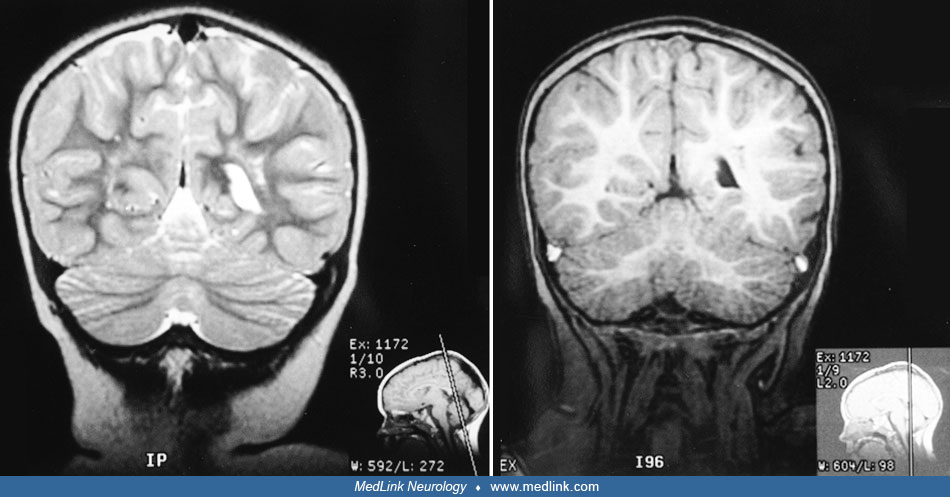

Methylmalonic acidemia
Jun. 01, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
In this article, the authors review the history of the discovery of phenylketonuria, one of the oldest known inborn errors of metabolism, and review the development of dietary treatment and neonatal screening for this disorder. The pathophysiologic mechanisms that are thought to cause central nervous system damage are discussed. New FDA-approved treatments and treatments in trial are revolutionizing the treatment of phenylketonuria in the 21st century.
|
• Phenylketonuria is caused by deficient activity of phenylalanine hydroxylase, an enzyme in the intermediary metabolism of the amino acid phenylalanine, and is one of the most common inborn errors of metabolism. | |
|
• Phenylketonuria is inherited as an autosomal recessive disorder. | |
|
• Neonatal screening for phenylketonuria allows for the early detection and treatment of infants with the disorder. | |
|
• Dietary therapy of phenylketonuria is based on restriction of dietary phenylalanine intake and largely prevents the major manifestations of the disorder, including profound developmental disability and seizures, but there are difficulties with lifelong adherence to the diet that may lead to cognitive impairment, particularly problems with executive functioning. | |
|
• Novel therapeutic approaches for phenylketonuria include sapropterin dihydrochloride treatment of a subset of patients who are responsive to the drug, large neutral amino acid supplementation to block phenylalanine uptake into brain, and enzyme substitution therapy with polyethylene glycol-conjugated phenylalanine ammonia lyase. |
In 1934, Følling reported a compound in the urine of two developmentally disabled individuals that reacted with ferric chloride to produce a deep green color. He subsequently identified this compound as phenylpyruvic acid in the urine of 10 developmentally delayed individuals (16). Penrose and Quastel named the disease "phenylketonuria” and first attempted dietary therapy (39). The metabolic defect in phenylketonuria was determined to be impaired hydroxylation of phenylalanine to tyrosine (23), and the liver enzyme phenylalanine hydroxylase was ultimately shown to be deficient in individuals with phenylketonuria (24). With the development of a simple bacterial inhibition assay, known as the Guthrie test (19), and along with the successful treatment by dietary restriction of phenylalanine using synthetic diets (06), neonatal screening and early treatment were introduced. Most neonatal screening programs have now discontinued the use of the Guthrie test to detect elevated phenylalanine concentrations in favor of the faster, more sensitive tandem mass spectrometry method that also detects several other amino acidopathies. The human gene encoding phenylalanine hydroxylase was cloned in 1983 (62).
With the successful treatment of phenylketonuria, a group of individuals was identified who, despite dietary restriction of phenylalanine, had progressive neurologic disease and were designated as having "malignant phenylketonuria." These children were found to have a deficiency of one of the enzymes of tetrahydrobiopterin metabolism. Tetrahydrobiopterin is a necessary cofactor for phenylalanine hydroxylase, tyrosine-3-hydroxylase, and tryptophan-5-hydroxylase (the latter two are rate-limiting enzymes in the synthesis of catecholamines and serotonin, respectively), and all forms of nitric oxide synthase. In addition to having elevated phenylalanine, children with defects in the metabolism of tetrahydrobiopterin exhibit deficiency of biogenic amine neurotransmitters in the central nervous system. Treatment by dietary phenylalanine restriction alone is not successful, but must be combined with tetrahydrobiopterin replacement and administration of L-DOPA and 5-hydroxytryptophan to address the neurotransmitter deficiencies.
Nomenclature for this group of inborn errors of metabolism can be confusing because of the great phenotypic variability (45). The 2014 American College of Medical Genetics and Genomics guidelines on the diagnosis and management of phenylalanine hydroxylase deficiency recommend that the term “phenylalanine hydroxylase (PAH) deficiency” be used to describe all affected individuals who previously have been described as having phenylketonuria. In this review, we will use the historical term “phenylketonuria.” This supports the concept that there is a spectrum of phenylalanine hydroxylase deficiency. This guideline recommends a unifying nomenclature and, therefore, refers to the spectrum of phenylalanine hydroxylase deficiency, not specifically relying on the blood phenylalanine concentration; although the guidelines recognize that the most severe form is still likely to be referred to as “classical phenylketonuria” in many settings. Historically, those with the most severe phenylalanine hydroxylase deficiency had untreated blood phenylalanine concentrations typically greater than 1200 μmol/l (mean normal concentration: 60 µmol/l) and were called “classical phenylketonuria”. On the milder side of the spectrum, patients with untreated blood phenylalanine concentrations greater than normal concentrations, but less than 1200 μmol/l, were previously described as having hyperphenylalaninemia.
Nearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125