

Cockayne syndrome
May. 08, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Clinical sequencing studies continue to expand our knowledge of the diverse genetic landscape of single enzyme defects of peroxisomal beta-oxidation. As a result of clinical whole exome sequencing and whole genome sequencing, several new clinical subtypes with milder presentations have been discovered. Moreover, genetic analyses have provided novel insights into the molecular basis of observed heterogenous clinical phenotypes.
|
• Single enzyme defects of peroxisomal beta-oxidation reported in the clinic are acyl-CoA oxidase 1 (ACOX1), acyl-CoA oxidase 2 (ACOX2), D-bifunctional protein (HSD17B4), sterol-carrier protein X (SCP2), alpha-methylacyl-CoA racemase (AMACR), ATP-binding cassette transporter protein family D member 3 (ABCD3), and acyl-CoA binding domain containing protein 5 (ACBD5) deficiencies. | |
|
• Inherited deficiencies in the ABCD1, ABCD3, or ACBD5 peroxisomal membrane proteins can also impair peroxisomal beta-oxidation by compromising the import of relevant substrates into the peroxisome matrix for beta-oxidation. | |
|
• Single enzyme defects of peroxisomal beta-oxidation are rare genetic disorders that most typically have autosomal recessive modes of transmission. | |
|
• Most have highly variable degrees of severity that are influenced by any residual levels of enzymatic activity. | |
|
• Although typically affecting multiple organ systems, the majority of the single enzyme defects of peroxisomal beta-oxidation are characterized by neurodevelopmental and neurodegenerative phenotypes that can vary in severity. | |
|
• In addition to genetic profiling, biochemical analyses of peroxisomal metabolites are essential for establishing a diagnosis. |
Peroxisomes are membrane-bound metabolic organelles present in nearly all eukaryotic cell types and are involved in cell signaling processes (114; 112; 95). They were first described as "microbodies" in the electron-microscopic examination of mouse kidney cells (82). Later, they were named “peroxisomes” by Christian De Duve and Pierre Baudhuin (21). Goldfischer and colleagues first established an association between the presence and functions of peroxisomes and human disease (40). Ensuing investigations over the next five decades led to the discovery of numerous connections between peroxisome dysfunction and the development and progression of rare and common disorders.
Peroxisomal disorders with Mendelian inheritance patterns can be broadly subdivided into those involving (i) defects in peroxisome assembly or (ii) the functions of single enzymes or membrane transporters. The former are currently referred to as peroxisome biogenesis disorders. Peroxisome biogenesis disorders, such as Zellweger spectrum disorder and rhizomelic chondrodysplasia punctata type 1 (RCDP1), are caused by biallelic loss-of-function variants in PEX genes encoding proteins involved in peroxisome biogenesis called peroxins (60). As a result, peroxisomes are improperly assembled and often diminished in number, which results in impaired metabolic functions that affect multiple organ systems (52; 11).
There are several different Mendelian disorders associated with defects in single peroxisome enzymes or membrane transporters. For example, loss-of-function variants in ABCD1, a peroxisomal transmembrane protein that transports very long-chain fatty acids (VLCFA, containing 22 or more carbons) as CoA-esters into the peroxisome where they are catabolized by peroxisomal beta-oxidation, causes the most frequent peroxisomal disorder, X-linked adrenoleukodystrophy (45). Furthermore, there are Mendelian disorders in which peroxisomes are present, but distinct enzymes normally located in their matrix are absent or defective. They are referred to as peroxisomal single enzyme defects (117; 106). Given the wide variety of peroxisomal metabolic functions, single enzyme defects of peroxisomal beta-oxidation can be further classified according to their impact on distinct metabolic pathways and processes. Single enzyme defects of peroxisomal beta-oxidation are the focus of this article. Although most early studies focused on patients with the most severe disease, more recent clinical sequencing has identified longer-surviving patients with hypomorphic alleles that convey residual enzymatic activity and can lessen disease severity.
Lazarow and de Duve were the first to provide evidence of a peroxisomal fatty acyl-CoA oxidizing system (56). It is now known that biallelic loss-of-function variants in any of a group of proteins required for peroxisomal fatty acid beta-oxidation can lead to disease (114; 117; 113; 106). Mendelian disorders caused by inherited deficiencies in enzymatic activities directly involved in peroxisomal beta-oxidation include acyl-CoA oxidase 1 and 2 (ACOX1/2), D-bifunctional protein (HSD17B4), alpha-methylacyl-CoA racemase (AMACR), and sterol-carrier protein X (SCP2). Single enzyme defects of peroxisomal beta-oxidation are relatively understudied disorders. As such, many aspects, such as variations in the initially described phenotypes, the natural history of disease, the characterization of the underlying genetic defects, and the evolution of the clinical manifestations from these genetic defects, are still under investigation.
To date, all reported cases of peroxisomal acyl-CoA oxidase deficiency are due to loss-of-function variants in the ACOX1 or ACOX2 genes (117). ACOX1 is involved in the peroxisomal catabolism of straight-chain substrates, including VLCFAs, dicarboxylic acids, and polyunsaturated fatty acids (31). Poll-The and colleagues were the first to report acyl-CoA deficiency in the case of two siblings who began their neonatal period with severe muscle hypotonia and seizures (79). Their shared features included delayed psychomotor development and eventual progressive neurologic regression, sensorineural hearing deficits, and an abnormal electroretinogram. Although they showed progressive leukodystrophy, there were no signs of cortical malformation. By 2021, over 30 patients with ACOX1 deficiency with variable ranges of severity had been reported (67). Since then, an additional three patients with ACOX1 deficiency have been reported (05). Severe early-onset ACOX1 deficiency can involve neonatal hypotonia, seizures, progressive leukodystrophy, impaired vision and hearing, hepatomegaly, severely delayed psychomotor development, and a shortened lifespan (28; 12; 117). More moderately affected siblings that showed mild global developmental delay from infancy have subsequently been reported (67). They began to regress at 5 years of age and gradually manifested with cerebellar ataxia, dysarthria, pyramidal tract dysfunction, and dysphasia (67). An even milder form has been described in adult siblings, 52 and 55 years of age, both of whom were nonambulatory, with mild to moderate cognitive impairment and impaired vision and brain MRI showing profound atrophy of the brainstem and cerebellum in both patients (26). Subsequently, three patients with the same de novo dominant gain-of-function ACOX1 p.N237S variant were described (17). All three presented with a progressive myeloneuropathy and sensorineural hearing loss; onset ranged from 3 to 12 years of age (17). The authors proposed naming the disease caused by the de novo gain-of-function ACOX1 pathogenic variant Mitchell disease in honor of the first patient studied. It is also referred to as Mitchell syndrome (https://www.omim.org/entry/618960).
In contrast to ACOX1, ACOX2 is involved in the peroxisomal catabolism of CoA-esters of branched-chain fatty acids (BCFAs) and C27-bile acid intermediates (31). Only four cases of ACOX2 deficiency have been reported to date (103; 66; 31; 127). The most severely affected patient displayed seizures 2 days after birth and died before 1 year of age (31). The authors noted the patient was born from a consanguineous union and that additional genetic causes could not be excluded. A more moderately affected 8-year-old child showed intermittently elevated transaminase levels, liver fibrosis, elevated bile acid intermediates, slurred speech, vertical gaze palsy, slight dysmetria, and mild gait ataxia as well as mild cognitive impairment (103; 127). Two mildly affected siblings (13 and 16 years old) showed hypertransaminasemia that peaked on exposure to drugs, such as nonsteroidal anti-inflammatory drugs (NSAIDs) and D-penicillamine in one and anti-flu medication in the other (66).
D-bifunctional protein deficiency (also called DBP deficiency or HSD17B4 deficiency) is the most frequently diagnosed single enzyme defect of peroxisomal fatty acid beta-oxidation, with over 130 cases reported in the literature to date (14). DBP is caused by biallelic loss-of-function variants in the HSD17B4 gene that encodes 17-beta-estradiol dehydrogenase, often referred to as D-bifunctional protein (109). DBP/HSD17B4 is involved in the catabolism of VLCFAs, BCFAs, and bile acid intermediates (29). Watkins and colleagues were the first to report a patient with a peroxisomal bifunctional enzyme deficiency (120). Although this initial report claimed the patient lacked L-bifunctional protein, it was later established that they lacked D-bifunctional protein (98).
Wanders and colleagues divided patients with DBP deficiency into three subgroups based on the extent of their enzyme deficiency (107). D-bifunctional protein encompasses two primary enzyme activities: enoyl-CoA hydratase and 3-hydroxyacyl-CoA dehydrogenase (107). Type I patients lack both activities, whereas type II patients lack only hydratase activity, and type III patients lack only dehydrogenase activity. In a series of 110 severely affected patients typically surviving less than 36 months, individuals with types I, II, or III DBP deficiency were reported (29c). The results suggested that the amount of residual DBP activity correlates with the severity of medical phenotypes and that a genotype-phenotype correlation exists. Adolescent brothers with milder symptoms were found to have a missense variant in the hydratase domain of one allele and a missense variant in the dehydrogenase domain of the other allele, suggesting they represent a new classification (type IV) (65). Since 2017, numerous reports have emerged of severe (07; 14; 55; 84; 16; 126; 123), moderate (25; 47; 55; 05; 125), and milder (15; 62; 61) forms of DBP deficiency.
Ferdinandusse and colleagues reported the first cases of AMACR deficiency (27; 36; 64). Two patients presented with adult-onset sensory motor neuropathy. One also had epileptic seizures and pigmentary retinopathy, whereas the other patient had upper motor neuron signs in the legs. A third patient was a child who was diagnosed with Niemann-Pick disease type C (NPC) complementation group 1 and showed progressive neurologic signs at 18 months of age. Since then, additional adult patients (18; 50; 89; 23; 91; 43) and juvenile-onset cases (100; 86; 94; 03; 53) have been described in the literature.
To date, there has been only one report of a patient with a sterol carrier protein X (SCPx, now named SCP2) deficiency (29b). In this case, the patient was homozygous for a frameshift variant in the SCP2 gene. SCPx has an amino-terminal thiolase domain and a carboxy-terminal sterol carrier protein 2 (SCP2) domain, and after SCPx protein import into the peroxisomal matrix, a proteolytic processing event gives rise to two separate proteins with thiolase (important for peroxisomal beta-oxidation) and SCP2 activities, respectively. The resulting thiolase can catabolize straight chain VLCFAs, BCFAs, and bile acid intermediates (117). The patient first experienced neurologic symptoms, including encephalopathy with dystonia as well as motor and peripheral neuropathies, in the second decade of life.
In 1986, Goldfischer and colleagues reported a patient whose clinical presentation and course resembled that of severe Zellweger spectrum disorder (39). Peroxisomes were present in their liver tissue, which ruled out a severe Zellweger spectrum disorder. Additional studies at that time suggested that the peroxisomal beta-oxidation enzyme 3-ketoacyl-CoA thiolase (ACAA1) was absent in this patient's liver (85). Later, it was found that peroxisomal 3-ketoacyl-CoA thiolase was present in postmortem brain, whereas D-bifunctional protein was absent (37). Pathogenic variants in the HSD17B4 gene were found and a diagnosis of DBP deficiency was made. The authors concluded that there is no longer evidence for the existence of ACAA1 peroxisomal thiolase deficiency as a distinct clinical entity (37).
Lastly, genetic deficiencies have been noted in three peroxisome membrane proteins that are transporters critical for normal peroxisomal beta-oxidation (ie, ABCD3, ACBD5, and ABCD1 deficiencies). The latter is the cause of the most common peroxisomal disorder, X-linked adrenoleukodystrophy, which is the subject of a separate review. Although there are a limited number of patients reported with ABCD3 and ACBD5 deficiencies, they are discussed in this review.
To date, there has been one report of a patient with an ABCD3 deficiency (34). ABCD3 is an abundant peroxisome membrane protein that facilitates entry of dicarboxylic acids, BCFAs, and bile acid intermediates into the peroxisome matrix for metabolism (34; 80). ABC (ATP-binding cassette) protein family members are involved in active transport and can have ATPase activity; thus, ABCD3 is included as a single enzyme defect of peroxisomal beta-oxidation. A patient presented with jaundice at 6 months and was hospitalized at 1.5 years of age with fever, gastroenteritis, hepatosplenomegaly, and severe anemia (34). By 4 years of age, the patient deteriorated rapidly with liver cirrhosis and hepatopulmonary syndrome and died 5 days after a liver transplantation.
To date, seven patients with a deficiency in ACBD5, a protein required for normal peroxisomal beta-oxidation, have been reported (32; 124; 09; 41). ACBD5 is a peroxisomal membrane protein with a membrane-spanning region and a cytosolic acyl-CoA binding domain proposed to sequester very long-chain fatty acyl-CoAs (ie, activated VLCFAs) in the cytosol and facilitate their transport into the peroxisomal matrix for catabolism (32; 124; 48). Although it has never been reported to have intrinsic enzymatic activity, it is mentioned in this review due to its role in peroxisomal beta-oxidation. ACBD5 also plays an important role in inducing contact site formation between peroxisomes and the endoplasmic reticulum (20). The first report of a putative ACBD5 deficiency involved three affected siblings with cone-rod dystrophy and psychomotor delay associated with significant white matter involvement (01). Later, a patient with ACBD5 deficiency who was born with a cleft palate and, at 7 months of age, showed a retinal dystrophy was reported (32). By 9 years of age, she showed a progressive leukodystrophy and ataxia. She was confirmed to be homozygous for a ACBD5 frameshift variant and showed elevated VLCFAs in blood and cultured skin fibroblasts. Since then, two siblings (41) and one adult with ACBD5 deficiency have been reported (09).
|
• The severity of single enzyme defects of peroxisomal beta-oxidation are influenced by any residual levels of enzymatic activity. | |
|
• Although single enzyme defects of peroxisomal beta-oxidation typically affect multiple organ systems depending on the gene involved, most are characterized by neurodevelopmental and neurodegenerative disease phenotypes that can vary in severity. | |
|
• Biochemical analyses of peroxisomal metabolites provide powerful means for establishing a diagnosis. |
All known single enzyme defects of peroxisomal beta-oxidation have the potential to have a clinical impact on the nervous system. Several genetic subtypes, such as ACOX1, DBP, and AMACR deficiencies, of which at least 20 patients of each subtype have been reported in the literature, are known to display variable expressivity. For such complex circumstances, early-onset forms of disease (defined herein as occurring during infancy) that are more severe separate from newly recognized, milder, adult-onset cases are discussed. In other subtypes (eg, ACOX2, ACBD5, and SCPx deficiencies) where limited (N< 5) numbers of patients preclude definitive conclusions, such subclassifications will not be made.
Early-onset ACOX1 and DBP deficiencies. These are most typically severe disorders that often cannot be readily distinguished from one another based on clinical presentations alone (Table 1). Their shared hallmark symptoms are muscle hypotonia and seizures, which present in the neonatal period. Severely affected individuals show severe delay in psychomotor development, and death typically occurs in infancy or early childhood. The clinical presentation in these disorders resembles that seen in patients with severe Zellweger spectrum disorder (90). Nevertheless, the clinical presentations of early-onset ACOX1 deficiency have been reported to be less severe than those of typical early-onset DBP deficiency (and, by extension, severe Zellweger spectrum disorder) because these patients can acquire some gross and fine motor skills (28; 90). Regardless, heterogeneous clinical presentations (Table 1), likely influenced by potential low-level residual ACOX1 or DBP activities in some individuals, make it challenging to make definitive statements that encompass all patients with early-onset ACOX1 or early-onset DBP deficiency.
ACOX1 deficiency–related data in Table 1 were adapted from (28). A cohort of 22 patients were included in this study, and data were reported based on patients for whom the indicated clinical presentations were evaluated. DBP deficiency–related data in Table 1 were adapted from (29). A cohort of 126 patients were included in this study, and data were reported based on patients for whom the indicated clinical presentations were evaluated.
|
Clinical presentation |
ACOX1 deficiency |
DBP deficiency |
|
Delayed maturation of white matter before 1 year |
17/48 | |
|
Delayed maturation of white matter after 1 year |
5/14 | |
|
Demyelination, cerebellar hemispheres |
3/47 | |
|
Demyelination, cerebral hemispheres |
9/47 | |
|
Craniofacial dysmorphism |
5/10 |
53/79 |
|
Failure to thrive |
3/8 |
27/61 |
|
Hepatomegaly |
5/10 |
32/73 |
|
Impairment/loss of hearing |
10/13 |
29/64 |
|
Impairment/loss of vision |
3/8 |
21/61 |
|
Loss of motor achievements |
10/12 |
7/61 |
|
Mean age at death |
5 years (range 4–10 years) |
Type 1: 6.9 months |
|
Neonatal hypotonia |
12/13 |
83/85 |
|
Seizures |
10/11 |
79/85 |
|
Visual system: nystagmus, strabismus, or failure to fixate objects at 2 months |
7/9 |
40/73 |
|
White matter abnormalities |
12/12 |
DBP deficiency can be subdivided into types I through III depending on the enzyme activity compromised. In an early report, the clinical presentations of patients with early-onset DBP deficiency did not significantly differ among types I through III (29). In nearly all early-onset patients, hypotonia was present in the neonatal period; it is usually generalized and severe. This can result in abnormal posture with limb abduction. Other signs of hypotonia reported in these patients are complete head lag (39), nuchal hypotonia (19), and weak neck muscles (08). In most cases, neonatal reflexes and Moro response are absent (39; 120; 59; 109); only Barth and associates reported the presence of a Moro response in their patient (08). Assessment of deep tendon reflexes shows variable results covering a spectrum from normal (79), through decreased (72; 120), to absent (39; 59; 109). Some antigravity movement, spontaneous or with stimulation, may be observed in a subset of patients. In patients who survive into infancy and childhood, hypotonia can be replaced by hypertonia with pyramidal tract dysfunction (79) or, depending on the cerebral progression of the disease, decerebrate posturing (08). Several authors report poor suck reflex and feeding difficulties requiring special feeding techniques, gavage feedings, or placement of a gastrostomy tube for long-term management (39; 72; 08; 59; 109).
Patients with early-onset ACOX1 and DBP deficiencies frequently present with distinctive craniofacial features (39; 72; 59; 109; 92; 29; 28; 14). In their large series, Ferdinandusse and colleagues reported that 68% of patients with DBP deficiency had dysmorphic features that resembled those found in patients with severe Zellweger spectrum disorder (29). Although some show only mildly distinctive features, such as a high forehead (72), one patient had profound craniofacial abnormalities, including a high forehead, epicanthal folds, low and broad nasal bridge, anteverted nostrils, micrognathia, and a high arched palate (109). One of the patients described by Mandel and colleagues had overall coarse features, a high forehead, shallow orbital ridges, and a long philtrum (59). One survey reported that half of patients with ACOX1 deficiency had craniofacial dysmorphism (28).
Patients with early-onset ACOX1 or early-onset DBP deficiencies typically manifest seizures in the neonatal period, with the age of onset varying between 15 minutes of life and 7 days (39; 19; 72; 79; 120; 08; 108; 29; 28; 73). Most of these episodes can be classified as multifocal and focal clonic seizures (104). Convulsions continue beyond the neonatal period, mainly as focal seizures, sometimes generalized (72; 08). In most patients, the seizures remain unresponsive to, or are only poorly controlled with, anticonvulsants. Electroencephalograms show frequent multifocal spikes (120), epileptiform discharges (79), and burst suppression patterns (59). One patient showed multifocal spike discharges and an electrographic focal seizure and, later in the course, lack of maturation and multifocal seizure activity, most prominent in slow-wave sleep (39).
An initial report of patients with early-onset ACOX1 deficiency stated that they generally achieved more motor milestones than those with early-onset DBP deficiency (121). In patients with early-onset DBP deficiency cases, overall psychomotor development was delayed, with virtually no developmental advances in the most severely affected individuals (39; 59). These findings were supported by later comparative studies involving larger numbers of patients (28).
In people with early-onset ACOX1 and early-onset DBP deficiencies, delays in psychomotor development and cognitive development may be exacerbated by hearing and visual impairment (39; 19; 79; 120; 08; 108; 29; 28). Over half of patients with early-onset ACOX1 or early-onset DBP deficiency showed evidence of vision abnormalities (eg, nystagmus, strabismus, or failure to fixate objects at 2 months of age), and a progressive loss of vision and hearing was noted in many patients (29; 28). Hearing is frequently reported to be impaired in patients with early-onset ACOX1 and early-onset DBP deficiencies (Table 1). Brainstem auditory evoked responses were normal shortly after birth in one patient (79), but this patient had sensorineural hearing loss later in life. In multiple patients, visual evoked responses and electroretinograms were also abnormal. Funduscopic exam was unremarkable in some patients early in life (120; 08; 59), but abnormalities may develop later. Nonspecific retinopathy (72), abnormal retinal pigmentation (08; 109), and retinal degeneration (79) have been described. Optic atrophy was present in one child with ACOX1 deficiency (79). Myopia and hyperopia have been reported (79), and cataracts were present in one patient (109).
Hepatomegaly has been noted in some people with early-onset ACOX1 or early-onset DBP deficiencies (39; 72; 79; 108; 59; 92; 29; 28) but seems to be mild and without significant impairment of hepatic function in many. Evidence of liver disease, such as fibrosis or steatosis, was found in about one fourth of patients with early-onset DBP deficiency (29). Liver function tests are typically normal or only mildly elevated. Symptomatic cirrhosis or hepatic failure has never been reported. In some patients, the hepatomegaly resolved after the first few months of life (19). Mild splenomegaly has been described in two patients (39; 79).
Individuals with early-onset ACOX1 or early-onset DBP deficiencies are either known (DBP) or predicted (ACOX1) to have an elevated risk of developing adrenal dysfunction (115; 14). Postmortem examination has revealed adrenal atrophy in some people with early-onset DBP deficiency (39; 19; 120; 29). Adrenal stimulation tests of people with early-onset DBP deficiency have been reported to be normal (08) or abnormal (72). Two siblings (born of a consanguineous union) with early-onset DBP deficiency and adrenal insufficiency were reported (14). Both showed classic manifestations of disease, including hypotonia and seizures. The younger sibling died at approximately 4 years of age, whereas the older sister was still living (7 years of age) but was encephalopathic and gastrostomy tube– and ventilator-dependent at the time of publication. In some patients with acyl-CoA oxidase deficiency (presumably ACOX1 deficiency), plasma corticotropin was elevated and cortisol was decreased (79). Although predicted based on the nature of the compromised enzymatic pathway, there are no reports of adrenal insufficiency in people with early-onset ACOX1 deficiency, but that may be influenced by the relatively small number of patients reported to date.
A delay of bone maturation, skeletal malformations, and calcific stippling has been noted in some cases of DBP deficiency, presumably all early onset (30). In initial reports of DBP deficiency, it was noted that patients with prolonged survival may have delayed bone maturation (19; 120). Late closure of the large fontanelle, presence of a metopic suture, and macrocephaly have been reported in people with early-onset DBP deficiency (19; 72; 109). Cranial dysmorphism has been reported in five of 10 patients with ACOX1 deficiency, but the dysmorphic features were milder than those observed in people with early-onset Zellweger spectrum disorder (28).
Cardiac malformations have been associated with early-onset DBP deficiency, including ventricular septal defects (39; 72). One of these patients developed intermittent cardiac failure and required treatment with diuretics and digoxin (39). Nevertheless, it is unclear if these individuals had an unreported comorbidity that influenced their cardiac phenotypes. Less is known about cardiac involvement in ACOX1 deficiency; however, this may be due to the relatively small number of patients reported to date.
Watkins and colleagues compared the clinical features of six patients with ACOX1 deficiency and 15 with DBP deficiency (121). Although patients with DBP deficiency had evidence of neuronal migration defects, patients with ACOX1 deficiency developed a progressive leukodystrophy between 2 and 3 years of age. White matter abnormalities were found in all patients with ACOX1 deficiency reported by Ferdinandusse and colleagues (28). In contrast, these authors reported delayed maturation of white matter in about one-third of patients with DBP deficiency but found white matter abnormalities in less than 20% of cases (29). Khan and colleagues reported serial MRI findings in a boy with type III DBP deficiency who survived 8 years 11 months (51). At 2 years 9 months, his MRI showed posterior white matter changes with involvement of the splenium of the corpus callosum, cerebellum, and brainstem, findings that are more typically seen in X-linked adrenoleukodystrophy. MRIs at 4, 5, and 6 years of age showed posterior to anterior progression of leukodystrophy, also more typical of X-linked adrenoleukodystrophy than of DBP deficiency. Excluding this unusual patient, the average age at death was 17.6 months for early-onset DBP deficiency and 5 years for early-onset ACOX1 deficiency.
In summary, early-onset ACOX1 and early-onset DBP deficiencies typically share many common features with severe Zellweger spectrum disorder, including hypotonia and seizures in the neonatal period. Nevertheless, there is variable expressivity in these patient populations. Specific disorders of peroxisomal dysfunction should be investigated in any patient with hypotonia and seizures, especially when combined with dysmorphic features, hearing deficit, visual impairment, and mild hepatomegaly.
Later-onset ACOX1 deficiency. The first reported diagnoses of ACOX1 deficiency in an adult involved 52-year-old male and 55-year-old female siblings, born of a consanguineous union, who were evaluated for mildly impaired cognitive function (26). For the male patient, early developmental milestones occurred in a typical timeline but showed scoliosis and a “clumsy right hand” at 10 years of age and a progressively unsteady gait until he became wheelchair-bound at 28 years of age. Neurologic examination was remarkable for slurring dysarthric speech, jerky head tremor, dystonic posturing of the arms, and ataxia. Spasticity was present in the lower extremity; reflexes were brisk but symmetrical. There was bilateral ankle clonus, and extensor plantar responses were elicited. His ophthalmologic examination revealed bilateral retinitis pigmentosa, decreased visual acuity, and gaze nystagmus. The 55-year-old sister had very similar clinical manifestations; however, her cognition was more impaired. Bilateral cataracts prevented visualization of her optic fundi. She was unable to move her legs but had normal reflexes. MRI findings in these adult siblings showed profound atrophy of the cerebellum and brainstem (particularly the pons in the male patient) but only modest cerebral atrophy. These findings differ significantly from the white matter changes seen in people with early-onset ACOX1 deficiency.
Later-onset DBP deficiency. In the last decade, multiple adolescents and adults with milder DBP deficiency phenotypes have been reported. An adolescent male, 16.5 years of age at the time of the study, had moderate-to-severe sensorineural hearing impairment since 3.5 years of age (65). He was otherwise healthy until 11 years of age when he developed progressive gait ataxia, which then required him to use a wheelchair except for short distances. He had bilateral pes cavus and mild hammertoe foot deformity, and diffuse areflexia, and his plantar reflex was flexor. A progressive sensorimotor polyneuropathy with demyelinating features was revealed by nerve conduction studies. Subclinical retinitis pigmentosa was reported. MRI at 12 years of age revealed cerebellar atrophy. There was no evidence of growth retardation, and he had age-appropriate pubertal development. The patient’s brother, 14 years of age when reported, also had moderate-to-severe sensorineural hearing impairment since he was 2 years of age (65). He had normal physical activity, but examination revealed very mild pes cavus and hyporeflexia as well as flexor plantar responses. Coordination was normal, but nerve conduction studies revealed a mild sensorimotor polyneuropathy with demyelinating features. Although visual acuity remained normal, peripheral retinal atrophy was noted. Although growth was normal, there was no evidence of pubarche at 14 years of age. Exome sequencing revealed both siblings were compound heterozygous for deleterious variants in the HSD17B4 gene. These patients had a missense variant in the hydratase domain of one allele along with a missense variant in the dehydrogenase domain of the other allele, prompting McMillan and colleagues to suggest that they represent a new classification of DBP deficiency (type IV).
A 35-year-old adult was reported to have had progressive ataxia since childhood and required a wheelchair at 29 years of age (57). He had mild developmental delay with cognitive impairment, and his medical record indicated documented azoospermia. Mild sensorineural hearing loss was detected at 34 years of age, and abnormalities were found on oculomotor examination. Progressive cerebellar volume loss was seen on MRIs obtained between 14 and 35 years of age. Although suspected of having a mitochondrial disorder, exome sequencing revealed the patient carried a rare HSD17B4 missense variant at a highly conserved residue along with a 12-kb deletion of exons 10 to 13. This confirmed a diagnosis of DBP deficiency.
Three adult siblings were also identified as having DBP deficiency by exome sequencing as standard biochemical profiling of blood samples did not provide a specific diagnosis (58). Although they ranged from 35 to 39 years of age at the time of the last report, hearing loss was noted in childhood (age range: 5 to 10 years), and ataxia was noted a few years later (age range: 10 to 14 years). Despite juvenile onset, disease progression was slowly progressive in nature. The authors reported that their clinical phenotypes included cerebellar atrophy and ataxia, intellectual decline, hearing loss, hypogonadism, hyperreflexia, and demyelinating sensory neuropathy (58). Furthermore, in two of the three patients, supratentorial white matter changes were observed (58).
Perrault syndrome (also known as XX gonadal dysgenesis) is a rare genetic disorder characterized by sensorineural deafness in females and males as well as ovarian dysgenesis in females (15). Neurologic features have also been reported in patients. Several genetic causes have been reported, including deleterious variants in the HSD17B4, HARS2, CLPP, and LARS2 genes (15). DBP deficiency was first reported in two sisters, 27 and 16 years of age at the time of publication, diagnosed with Perrault syndrome (78). Both showed short stature, mild intellectual disability, and progressive peripheral neuropathy. Genetic analyses revealed that both patients had biallelic loss-of-function alleles in the HSD17B4 gene. The older sister had cerebellar manifestations, including ataxia, dysarthria, and intention tremor; her MRI showed moderately severe atrophy of the cerebellar hemispheres and vermis. Ovarian dysgenesis has not been reported in DBP deficiency, primarily because few if any female patients have survived to puberty. Amor and colleagues described five new patients with “juvenile peroxisomal D-bifunctional enzyme deficiency,” and they summarized the clinical findings in 14 patients from eight families (04). Patients with adolescent and adult HSD14B deficiency, as well as Perrault syndrome, were considered as one group in this report. Chen and colleagues described six patients (from four families) with Perrault syndrome (15). Despite pathogenic HSD17B4 variants in either the enoyl-CoA hydratase domain, the hydroxyacyl-CoA dehydrogenase domain, or both, these patients appear to have normal VLCFA levels.
In the past 3 years, there have been multiple reports of moderate (25; 55; 05; 125) and milder (22; 62; 61) forms of DBP deficiency. These reports reinforce the concept of the clinical heterogeneity in the patient population that is, in part, influenced by the nature of the sequence variant and residual enzymatic activity. As such, the assumptions of disease severity based solely on knowledge of the causal gene should not be made.
Sterol carrier protein X (SCP2) deficiency. The first report of a patient with deficiency of sterol carrier protein X (SCP2) was made in 2006 (35). The patient, diagnosed at 45 years of age, had a 28-year history of spasmodic torticollis and dystonic head tremor. At age 44, he was evaluated due to worsening dystonic symptoms. Neurologic findings at that time included hyposmia, pathological saccadic eye movements, brisk deep tendon reflexes of the upper extremities but diminished lower extremity reflexes, plantar sole responses, reduced vibration sense, slight cerebellar ataxia with intention tremor, and balance and gait impairment. No pareses were present, and his superficial sensation was normal. Ophthalmological examination was unremarkable. MRI revealed bilateral hyperintense signals in the thalamus, butterfly-like lesions in the pons, and lesions in the occipital region. Nerve conduction studies of both the upper and lower extremities were abnormal. The patient was infertile, with hypergonadotropic hypogonadism and azoospermia. He had two male siblings, one (1 year younger) of whom was said to have similar symptoms; the other brother (4 years younger) was reportedly asymptomatic.
AMACR deficiency. To date, 17 individuals with AMACR deficiency have been reported. Patients’ ages at diagnosis ranged from about 1 to 57 years at the time of publication. Despite clinical heterogeneity, possibly related to patient age and levels of residual AMACR activity, some common clinical features are emerging (Table 2). The clinical presentation of this disorder in adults differs significantly from early-onset ACOX1 or DBP deficiency but is reminiscent of some neurologic aspects of adult Refsum disease (94; 118). Neurologic disease, including learning difficulties and encephalopathic episodes, sensory and motor neuropathy, seizures, and cerebellar signs, have been reported (03). In some patients, clinical manifestations included cholestatic liver disease or giant cell hepatitis, or both; fat-soluble vitamin deficiency; bloody stool in early life; and vitamin K deficiency. Finally, some patients develop recurrent rhabdomyolysis and stroke-like episodes, tremor, and hypogonadism. More recently, retinal dysfunction, even in the absence of visual symptoms, was highlighted as being possibly useful for clinical diagnosis and monitoring response to dietary interventions.
The first report of AMACR deficiency included two patients who presented with adult-onset sensory motor neuropathy (27). A patient, diagnosed at 44 years of age, had retinitis pigmentosa, primary hypogonadism, epileptic seizures, and a widespread axonal neuropathy affecting the legs more than the upper extremity. A more complete case report of this patient was subsequently published (64). The second adult patient, a female who did not develop symptoms until 48 years of age, developed a spastic paraparesis of the lower extremity, with normal MRI of the cervical spine (27). Nerve conduction studies revealed evidence of a demyelinating polyneuropathy.
Another patient, diagnosed with schizophrenia at 23 years of age, subsequently suffered from recurrent bouts of rhabdomyolysis and stroke-like episodes that eventually led to a diagnosis of AMACR deficiency about 11 years later (50). An MRI at the age of 23 showed T2-weighted increased signal in pons, thalamus, and cerebral peduncles; MRIs following stroke-like episodes at the ages of 33 and 34 showed cortical edema. He had decreased visual acuity and an abnormal electroretinogram without pigment clumping. Following his second stroke-like episode, a right frontal cortex biopsy was performed that showed discrete edema, endothelial cell hyperplasia, microglial activation, axonal ballooning, and reactive gliosis, consistent with acute ischemia without inflammation. The biochemical studies that eventually led to a diagnosis of AMACR deficiency were conducted because the first and third patients’ symptoms resembled those of adult Refsum disease; the second patient’s symptoms resembled those found in adrenomyeloneuropathy, the X-linked adrenoleukodystrophy phenotype that presents in symptomatic heterozygotes during late adolescence or adulthood.
Severe forms of AMACR deficiency have been diagnosed in children (Table 2). One child was also documented as having Niemann-Pick C disease (27). No symptoms directly related to AMACR deficiency could be detected due to the concurrent Niemann-Pick C disease. Another child presented at 2 weeks of age (100; 86). Her primary symptoms were consistent with vitamin K deficiency, and serum levels of all fat-soluble vitamins were low. The results of a liver biopsy were consistent with giant-cell neonatal hepatitis and severe cholestasis, prompting the biochemical analyses that led to a diagnosis of AMACR deficiency. An older sibling was said to have died of vitamin K deficiency. The sibling’s liver was used for orthotropic transplantation into a 28-month-old child with end-stage liver disease, and a posttransplantation biopsy was significant for evidence of acute rejection, bile duct proliferation, and fibrosis. Although unproven on the protein or DNA level, the sibling most likely also had AMACR deficiency.
|
Age at earliest symptomsa/sex |
Clinical findings (# of patients in reported group) |
References |
|
< 2-year-old M |
Seizures (1), encephalopathy (1), retinitis pigmentosa (1), sensory-motor neuropathy (3), migraine (1), hypogonadism (1), learning difficulties (1) |
(27; 64) |
|
< 1-year-old F |
Hematochezia (secondary to a coagulopathy from vitamin K deficiency), giant-cell neonatal hepatitis, cholestatic liver disease |
(100; 86) |
|
36-year-old F |
Seizures, encephalopathy, retinitis pigmentosa, cataract, tremor, cerebellar signs, neuropathy, depression, migraine |
(18) |
|
13-year-old F |
Seizures, encephalopathy, sensory-motor neuropathy, cognitive decline, depression, homonymous hemianopia |
(94) |
|
Early adult male |
Seizures, encephalopathy, retinitis pigmentosa, sensory neuropathy, hypogonadism, learning difficulties |
(89) |
|
23-year-old M |
Rhabdomyolysis, stroke-like episodes, seizures, encephalopathy, sensory neuropathy, degenerative retinopathy, schizophrenia |
(50) |
|
25-year-old M |
Seizures, encephalopathy, pigmentary retinopathy, low testosterone level |
(91) |
|
50-year-old M |
Seizures, cerebellar signs, sensory-motor neuropathy, decline in short-term memory |
(23) |
|
30-year-old M |
Seizures (2), encephalopathy (2), tremor (1), sensory-motor neuropathy (2), pigmentary retinopathy (2), cataract (2), type 2 diabetes (2) |
(43) |
|
2-year-old M |
Cholelithiasis (2), cholestatic liver disease (3), subtle retinopathy (3), fat-soluble vitamin deficiency (3), learning difficulties (3) |
(03) |
|
Early childhood M |
Oligophrenia, hypersexuality disorder, bipolar disorder with severe depression, rhabdomyolysis, gynaecomastia, electroencephalograms showed paroxysmal activity with epileptic discharges |
(53) |
|
a First noted appearance of any symptom | ||
|
Adapted from (03) | ||
ACOX2 deficiency. The first diagnosed case of ACOX2 deficiency was reported in an 8-year-old child who showed normal growth (103). Nevertheless, an evaluation at 8 months for vomiting, presumably secondary to acute gastroenteritis, revealed elevated transaminase levels. His transaminase levels were intermittently elevated over subsequent years. A liver biopsy at 6 years revealed many thin fibrous septa, swollen hepatocytes, glycogenated nuclei, and focal acinar transformation, but no obvious cholestasis or steatosis. He had mildly delayed language development and mild intellectual disability. Neurologic evaluation revealed slurred speech, vertical gaze palsy, slight dysmetria, and mild gait ataxia. His performance on the Wechsler Intelligence Test for Children was 66 (mild intellectual disability). Fundoscopy and brain MRI results were both normal. Exome sequencing revealed a homozygous nonsense variant in the ACOX2 gene. Consistent with a proposed role for ACOX2 in synthesis of bile acids from cholesterol via beta-oxidation of the aliphatic side chain, the patient’s plasma and urine contained elevated levels of C27 bile acid precursors and low to low-normal levels of mature bile acids. However, BCFA (ie, phytanic and pristanic) levels were normal. The authors noted that a trial of primary bile acid therapy, which reduces synthesis of potentially toxic intermediates, was planned for this patient, who was at least 6.5 years of age at the time of the report (103). A second patient with ACOX2 deficiency presented at 16 years of age with persistent hypertransaminasemia of unknown origin (66). A liver biopsy showed only mild intracellular cholestasis. The patient was treated with cholestyramine, which improved the hypertransaminasemia. Subsequent analysis of his bile acids revealed decreased C24 species and elevated C27 precursors, in both serum and urine.
ABCD3 deficiency. There is only one report of a patient with ABCD3 deficiency (34). The patient was born of a consanguineous union and showed normal early growth and development. Nevertheless, mild jaundice was noted during the neonatal period and around 6 months but was not treated. Abdominal distension was also observed at 1 year. While being treated at a hospital at 1.5 years for fever and gastroenteritis due to rotavirus infection, hepatosplenomegaly and paleness were noted. She was found to be anemic and mildly jaundiced and had elevated serum transaminases. The anemia was thought to be due to iron deficiency. A liver biopsy at 1.5 years revealed evidence of hepatocellular regeneration, but there was minimal intracanalicular and intracytoplasmic cholestasis. Fibrosis was present, but no necrosis or steatosis. At 2 years of age, she was started on ursodeoxycholate treatment. Despite normal growth and development, her liver and spleen enlarged further over the next 2 years, leading to pancytopenia. She developed severe portal hypertension around 3 years of age, again with mild jaundice. Plasma bile acid, and particularly bile acid precursor, levels were markedly increased. At 4 years of age, the patient rapidly deteriorated. Decompensated cirrhosis and hepatopulmonary syndrome necessitated a liver transplant. The patient died 5 days posttransplant from respiratory complications. Skin fibroblasts were obtained and found to have a reduced number of peroxisomes that also showed enlargement, similar to those seen in skin fibroblasts obtained from patients with ACOX1 or DBP deficiencies. The rates of VLCFA beta-oxidation in these cells were in the normal range, whereas the rates of pristanic acid beta-oxidation were reduced relative to controls. Sequencing analysis revealed the patient to be homozygous for a 1758-bp deletion in the ABCD3 gene, resulting in a deletion of exon 24 and part of the 3′-UTR at the cDNA level.
ACBD5 deficiency. A variant in the ACBD5 gene, which encodes a peroxisomal membrane protein with a putative acyl-CoA binding domain, predicted to abolish a consensus splice-donor site was detected in three affected siblings who presented with cone-rod dystrophy and psychomotor delay associated with significant white matter involvement (01). No further clinical information on these patients was available. The clinical picture of a fourth patient from a different family was reported in 2017 (32). She was the only affected of three siblings from healthy, consanguineous parents. She was healthy at birth except for a cleft palate. Abnormal eye movements were noted at 7 months, and retinal rod-cone dystrophy was diagnosed. She had delayed motor skill development and an unsteady gait. By 2 years of age, her gait had become progressively abnormal. By 4 years of age, her vocabulary was limited, and she was dysarthric despite normal hearing. She developed progressive microcephaly with facial dysmorphisms (including a tubular nose, hypotelorism, prominent ears, bilateral ptosis) and rotatory nystagmus. Motor dysfunction was marked, with a positive Gowers and proximal weakness; there was increased tone in her arms and legs. She had a wide-based gait with truncal titubation and waddling. By 9 years of age, she could walk only with two-handed assistance or short distances with a walker. Brain MRI at 4 years revealed hypomyelination with diffuse T2 signal abnormality in deep white matter, with relative sparing of the subcortical U fibers. Signal abnormality was also seen to involve the long tracts in the brainstem, including the pyramidal tracts, the medial lemniscus, and the inferior cerebellar peduncles (32). Due to mildly elevated VLCFA levels, peroxisomal dysfunction was suspected. The patient was found to be homozygous for a variant deleting exons 7 and 8 of the ACBD5 gene. In vitro studies in cultured patient skin fibroblasts confirmed that ACBD5 loss-of-function variants affect peroxisomal beta-oxidation of VLCFA (32). Skin fibroblasts from one of the original three patients were studied and found to have defective peroxisomal VLCFA beta-oxidation (124).
A 36-year-old patient with ACBD5 deficiency has been reported (09). Although otherwise healthy, she was diagnosed with nystagmus and optic atrophy at 1 month of age, and by the age of 3, displayed signs of developmental regression and progressive lower extremity weakness. The patient showed significant cognitive decline with rapidly progressive motor and speech regression and lost the ability to move her lower extremities by 11 to 13 years of age. Furthermore, she began to show upper extremity weakness and difficulty in fine motor movements at 15 years of age. At the time of publication, she had severe dysarthria and spastic paraparesis with contractures of both upper and lower extremities. The results of a recent brain MRI indicated supratentorial and infratentorial atrophy with diffuse white matter signal abnormalities, unchanged from a prior brain MRI at 29 years of age. Her ophthalmic evaluation revealed optic nerve pallor, attenuation of eye vessels, and diffuse granularity of retinal pigment epithelium bilaterally. Follow-up full-field electroretinography (FFE) confirmed that she has a severe cone-rod dystrophy.
Single enzyme defects of peroxisomal beta-oxidation are either known (ACBD5, ACOX1, ACOX2, AMACR, and DBP deficiencies) or predicted (ABCD3 and SCP2) to have variable severity based on the general principle that residual functions of the impaired protein of interest can attenuate clinical manifestations of disease. Therefore, it is not appropriate to assume a prognosis based simply on the name of the condition. Nevertheless, the prognosis for patients with early-onset ACOX1 or early-onset DBP deficiency is typically poor (Table 1). Patients with early-onset DBP deficiency have frequent seizures and show only minimal development, with the average age at death reported to be 17.6 months (121; 29). Nevertheless, patients with DBP deficiency who have survived longer than 2 years are known; all have type II or III enzyme deficiency. Longer survival best correlated with low VLCFA levels and a high rate of VLCFA beta-oxidation in fibroblasts (29). Adults with DBP deficiency showed either normal or only mildly elevated VLCFA levels (78; 65; 57; 58). Patients with early-onset ACOX1 deficiency are somewhat less profoundly affected, developing some milestones. Nevertheless, the average age at death is about 5 years (28). Given that only a few cases are reported, the long-term prognosis for ABCD3, ACOX2, ACBD5, and SCP2 deficiency remains unknown. Based on the current literature, almost all people with AMACR deficiency live beyond their fourth decade.
Early-onset ACOX1 deficiency. The patient, first seen at 15 months of age, was born after a full-term pregnancy to a 22-year-old G2P1 woman. Labor and delivery were normal. The patient weighed 7 pounds at birth, did well in the newborn period (including feeding), and went home with his mother at 2 days of age. At 2 months of age, he had three tonic-clonic seizures in one day. He was placed on carbamazepine for 2 months until his mother stopped treatment. The patient has had no further seizures and has had slow but steady development. At 9 months, he could sit by himself but not stand. He knew his mother from strangers and was socially interactive. His vision and hearing were normal. On physical examination, his weight was 20.9 pounds (10th to 25th percentile), height was 81 cm (75th percentile), and head circumference was 45.2 cm (10th percentile). He was thin with no unusual features, and the remainder of the general exam was normal.
Neurologic examination revealed that he was awake and alert, had a social smile, sought out his mother, and played with toys. His cranial nerves were intact. Motor examination revealed that he was markedly hypotonic with normal muscle mass. The patient slipped through when held in vertical suspension and arched his back when placed in ventral suspension. There was righting of his head in this position. He could transfer objects between hands, but when reaching for objects, there was a mild tremor and dysmetria. He could sit unsupported and could stand with support with his feet apart but was not interested in cruising. Sensation was intact for light touch and pain. Examination of cerebellar function revealed dysmetria, which appeared to be secondary to low tone. Reflexes were 1+ at the knees, biceps, and ankles, and 0 at the triceps and brachioradialis. His reflexes were symmetric, with no pathological reflexes noted, and his toes were down going.
Laboratory studies showed normal hematologic, liver, and adrenal functions. Plasma VLCFA analyses revealed that his C26:0 levels were mildly elevated at 0.51 µg/ml (normal mean 0.22 µg/ml). Magnetic resonance imaging demonstrated abnormal white matter signal involving the parietal occipital regions and centrum semiovale bilaterally. He began to manifest progressive loss of skills, including motor and visual function, and was last reported to have unresponsive wakefulness syndrome.
Early-onset DBP deficiency. The patient was born after a full-term pregnancy to a 19-year-old G1P0 woman and weighed 7 lbs 8 oz at birth. She required oxygen and had two seizures in the first day of life. Over the next few days, she began having frequent seizures and was noted to be hypotonic. She spent 1.5 months in the hospital and was only home for a few days, when she had to be readmitted for apnea and seizures. She spent the first 4 months in the hospital. A portion of the time in the hospital was complicated by pneumonia and time on the ventilator. She initially took a bottle for 2 months but then lost this ability with the onset of the above complications and required a gastrostomy tube.
When seen at 10 months, she had a full forehead with a small anterior fontanelle and a fine rash on her face and trunk. On eye examination, her sclerae were clear and the conjunctiva pink. The remainder of her general examination was unremarkable, with normal liver functions, no skeletal problems, and no bleeding abnormalities. Although she no longer had apnea, she still had daily seizures. She would turn her head to the side in a fencer posture and then had clonic movements. On physical examination, she was a sleeping infant who was difficult to arouse. When finally aroused, she had a 2-minute seizure on awakening. There was no visual tracking, although the pupils were reactive and extraocular movements were present. There were minimal corneal responses and diminished facial movement. There was no apparent response to voice. She had a decreased gag reflex. Her tongue was in the midline, and there were no fasciculations. Generally, there was minimal movement, and she was markedly hypotonic with decreased muscle mass. She grimaced to blood drawing. There were no reflexes, and her toes were nonreactive. Over the next few weeks, she continued to have uncontrolled seizures and died at eleven months of age.
The most typical underlying cause of single enzyme defects of peroxisomal beta-oxidation are biallelic genetic defects that result in a deficiency in the activity of a protein required for peroxisomal fatty acid beta-oxidation. Except for X-linked adrenoleukodystrophy, which is discussed in a different review, there is an autosomal recessive mode of transmission. Most typically, pathogenic variants result in loss-of-function alleles.
Except for some of the earliest published studies, pathogenic variants in most patients with single enzyme defects of peroxisomal beta-oxidation are documented. Ferdinandusse and colleagues described 20 distinct candidate deleterious sequence variants in 22 patients with ACOX1 deficiency (28). No clear-cut correlation of genotype with clinical phenotype was observed in early-onset ACOX1 deficiency. Two transcript variants of the human ACOX1 gene result from alternative splicing of two different exons 3 (28; 76). One patient with biochemical features typical of ACOX1 deficiency had a homozygous variant that deleted six amino acids encoded by exon 3II; this patient was still able to produce a functional ACOX1 that contained exon 3I (28). Exome sequencing has now identified several older patients with previously undiagnosed neurologic symptoms as having DBP deficiency. Several of these more mildly affected patients were compound heterozygous for deleterious HSD17B4 variants.
Before the HSD17B4 gene was discovered, it was thought that patients with a bifunctional protein deficiency had a L-bifunctional protein deficiency (EHHADH) (120). This discrepancy was resolved when Wanders and colleagues (98) found that the original “bifunctional enzyme deficiency” patient had deleterious variants in the HSD17B4 gene. These authors analyzed DNA from nine additional patients and found deleterious HSD17B4 variants. Subsequently, a large series of patients was investigated, and 61 different variants of interest were found in 110 patients (38). These investigators examined the available clinical and biochemical data for each patient along with the predicted effect of the variant on enzyme activity, protein folding, or protein dimerization. Deleterious variants identified in patients with a relatively mild clinical and biochemical presentation were found to be less detrimental to the protein structure than deleterious variants in severely affected patients. They concluded that the amount of residual activity of HSD17B4 correlated with the clinical severity. No case of EHHADH/L-bifunctional protein deficiency has been genetically confirmed.
Wanders and colleagues also cloned the human AMACR gene and found genetic variants of interest from their original three patients (27). This group later established a diagnosis of SCP2 deficiency in one patient and a diagnosis of ABCD3 deficiency in one patient by variant analysis (35; 34). An ACOX2 deleterious variant was found by exome sequencing (103).
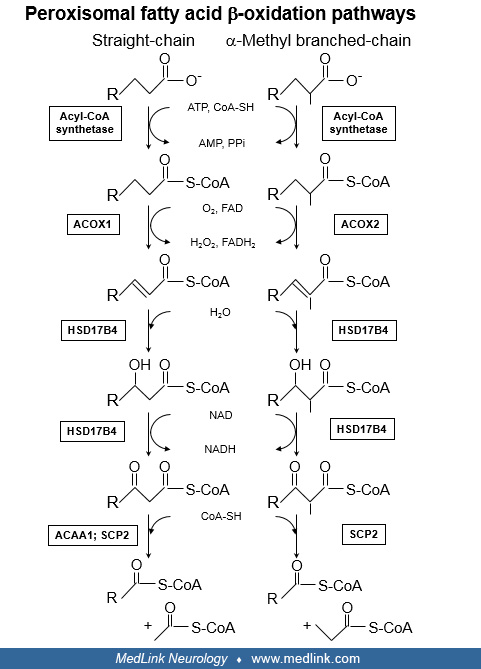
Primary functions of the peroxisomal fatty acid beta-oxidation pathway. The term “beta-oxidation” refers to the process by which an aliphatic carboxylic acid is shortened by cleavage between the second and third carbon atoms in the aliphatic chain. For a straight-chain fatty acid, one cycle of beta-oxidation yields a product shortened by two carbons plus acetyl-CoA. For fatty acids with a methyl branch on the second carbon, beta-oxidation yields a product shortened by three carbons plus propionyl-CoA.
In general, the shortened fatty acids can undergo additional cycles of beta-oxidation. However, complete degradation of fatty acids as seen in mitochondria does not occur in peroxisomes. The primary functions of the peroxisomal beta-oxidation pathway are: (1) to catabolize certain fatty acids that cannot be degraded by mitochondria and (2) to carry out key reactions in the synthesis of bile acids. The accumulation of substrates and metabolites proximal to a defective enzyme forms the pathogenesis and pathophysiology of the single enzyme defects in this pathway.
Some fatty acids are not oxidized by the traditional mitochondrial beta-oxidation pathway (used to degrade dietary and stored fatty acids for energy production) either because they cannot be transported into mitochondria, or they are not recognized as substrates by the mitochondrial enzymes. The physiologically significant fatty acids in this group are the very long-chain fatty acids (VLCFAs), phytanic acid (3,7,11,15- tetramethylhexadecanoic acid; a 3-methyl branched chain fatty acid), and pristanic acid (2,6,10,14-tetramethylpentadecanoic acid; a 2-methyl branched chain fatty acid).
VLCFAs originate from both the diet and endogenous synthesis. Phytanic acid is solely of dietary origin, and the presence of the 3-methyl branch prevents its degradation by beta-oxidation in either peroxisomes or mitochondria (122; 68). Pristanic acid is produced when phytanic acid is degraded by alpha-oxidation, a peroxisomal process in which the acid is shortened by one carbon. The methyl group in pristanic acid is now in the 2-position, allowing beta-oxidation to proceed. A primary defect in the alpha-oxidation pathway enzyme phytanoyl-CoA alpha-hydroxylase is the cause of adult Refsum disease, an adult-onset polyneuropathy (118). Because pristanic acid arises only from phytanic acid, defects in beta-oxidation that prevent pristanic acid degradation can slow down phytanic acid degradation, causing the latter to accumulate as well.
Bile acids are synthesized from cholesterol in a complex pathway involving several subcellular compartments. Modifications to the steroid nucleus and oxidation of one of the methyl groups in the side chain yield the precursors of primary bile acids. The precursor of cholic acid is trihydroxycholestanoic acid (THCA) and the precursor of chenodeoxycholic acid is dihydroxycholestanoic acid (DHCA). The terminal steps in bile acid synthesis involve shortening of the side chains of THCA and DHCA by three carbons. As discussed below, chain shortening is analogous to the first cycle of pristanic acid beta-oxidation. Both bile acid precursors and branched chain fatty acids contain asymmetric carbons; thus, these compounds exist in several stereoisomeric configurations.
Peroxisomal fatty acid beta-oxidation pathways and enzymes. The first step in fatty acid oxidation is activation by thioesterification to CoA, reactions catalyzed by specific acyl-CoA synthetases (119; 107).
Studies suggest thioesterification occurs outside the peroxisome. ACBD5 is a peroxisomal membrane protein with an acyl-CoA binding domain located in the cytosol; this protein is hypothesized to capture cytoplasmic VLCFA-CoA for transport into the peroxisome matrix (32; 124). CoA derivatives of VLCFA, branched-chain fatty acids, and bile acid precursors then enter the organelle via ATP-binding cassette proteins of the ABCD family. Specifically, saturated VLCFA-CoA are thought to require ABCD1 (the gene defective in X-linked adrenoleukodystrophy) for entry (99) and the CoA esters of phytanic acid, pristanic acid, and bile acid precursors enter via ABCD3 (34). Once inside the peroxisome, a double bond is inserted between carbons 2 and 3 in the aliphatic chain, a reaction catalyzed either by ACOX1 or by branched-chain acyl-CoA oxidase (ACOX2). ACOX1 acts on VLCFA and other straight-chain fatty acids, whereas ACOX2 acts on pristanic acid and the bile acid precursors. The next two enzymatic steps, hydration of the double bond (enoyl-CoA hydratase activity) and oxidation of the hydroxyl group to a keto group (hydroxy-acyl-CoA dehydrogenase activity), are both catalyzed by HSD17B4. Although it was initially thought that products of ACOX1 were processed by L-bifunctional protein (EHHADH) and products of branched-chain acyl-CoA oxidase were processed by HSD17B4, evidence from both biochemical and molecular genetic studies clearly indicate that HSD17B4 utilizes substrates generated by both ACOX1 and ACOX2. The precise role of EHHADH/L-bifunctional protein in peroxisomal fatty acid metabolism is currently uncertain. A study of genetically engineered mice suggested a role for EHHADH in bile acid synthesis, perhaps via an alternative pathway (30). Hepatocytes from EHHADH-deficient mice were shown to have reduced capacity to degrade dicarboxylic polyunsaturated fatty acids (75). Subsequent studies revealed that the increase in medium-chain (C6-C8) dicarboxylic acids in plasma and urine normally observed after fasting was virtually absent in EHHADH-deficient mice, confirming a role for EHHADH in the peroxisomal beta-oxidation of long-chain (C16-C18) dicarboxylic acids is disturbed (46).
Products of HSD17B4 or EHHADH (3-ketoacyl-CoAs) are then thiolytically cleaved in a CoA-dependent reaction. Peroxisomal thiolase (ACAA1) catalyzes this reaction for the 3-keto derivatives of VLCFA and other straight-chain fatty acids, but not for the 3-keto derivatives of pristanic acid and bile acid precursors. Thiolase activity was also found in amino terminal portion of a different peroxisomal protein, the 58-kDa sterol carrier protein X (SCP2); the carboxyl portion of this protein is identical to sterol carrier protein 2. SCP2 thiolase can accept 3-ketoacyl-CoA substrates from both straight-chain and branched-chain fatty acid catabolism, although it shows a preference for the latter. The products of ACAA1 and SCPX thiolase are the CoA derivatives of the chain-shortened substrates. In the case of VLCFA and pristanic acid degradation, these products can then go through several additional rounds of beta-oxidation, utilizing the same enzymes in a cyclic process. The other cleavage products of ACAA1 and SCPXT are the 2-carbon compound, acetyl-CoA (from VLCFA catabolism), or the 3-carbon compound, propionyl-CoA (from pristanic acid or THCA degradation).
Biochemical features of patients with deficiencies of peroxisomal beta-oxidation enzymes are summarized in Table 3.
|
Attribute |
ABCD3a |
Early-onset ACOX1 |
ACOX2 |
AMACR |
ACBD5b |
Early-onset DBP |
SCP2c |
|
Plasma C26:0 |
Normal |
Elevated |
Normal |
Normal |
Borderline-elevatedf |
Elevated |
Borderline-elevatedg |
|
Plasma phytanic acid |
Normal |
Normal |
Normal |
Normal to mildly elevated |
NR |
Normal |
Elevated |
|
Plasma pristanic acid |
Normal |
Normal |
Normal |
Elevated |
NR |
Elevated |
Elevated |
|
Plasma THCA; DHCA |
Elevated |
Normal |
Elevated |
Elevated |
NR |
Elevated |
Borderline-elevated THCAh |
|
Fibroblast C26:0 |
Borderline-reducedd |
Elevated |
NR |
NR |
Elevated |
Elevated |
Normal |
|
Fibroblast VLCFA beta-oxidation |
Borderline-reducede |
Reduced |
NR |
NR |
Reduced |
Reduced |
Normal |
|
Fibroblast pristanic acid beta-oxidation |
Reduced |
Normal |
Normal |
Reduced |
Elevated |
Reduced |
Reduced |
|
Fibroblast plasmalogen synthesis |
NR |
Normal |
NR |
NR |
NR |
Normal |
NR |
|
NR = not reported; borderline = marginal change from reported normal range | |||||||
|
a Data from (34) | |||||||
|
b Data from (32) | |||||||
|
c Data from (35) | |||||||
|
d Fibroblast C26:0 oxidation for the single patient with ABCD3 deficiency was 1247 pmol/(min mg), which was just outside the control range of 1273 to 143 pmol/(min mg). | |||||||
|
e Fibroblast C26:0 level in the single patient with ABCD3 deficiency was 0.17 umol/g, which was just outside the control range of 0.18 to 0.38 umol/g. | |||||||
|
f Plasma C26:0 level in the single patient with ACBD5 deficiency was 0.43 ug/ml, which was just outside the control range of 0.05 to 0.41 ug/ml. | |||||||
|
g Plasma C26:0 level in the single patient with SCP2 deficiency was 1.34 umol/l, which was just outside the control range of 0.46 to 1.31 umol/l. | |||||||
|
h Plasma DHCA level in the single patient with SCP2 deficiency was 0.1 umol/l, which was within the control range of 0.0 to 0.13 umol/l; Plasma THCA level in the single patient with SCP2 deficiency was 0.1 umol/l, which was just outside the control range of 0.0 to 0.09 umol/l. | |||||||
Patients with early-onset ACOX1 deficiency accumulate VLCFA but not phytanic acid, pristanic acid, or bile acid precursors (THCA and DHCA) (121; 74). Defects in VLCFA catabolism can clearly be demonstrated in cultured skin fibroblasts (121). In contrast, patients with early-onset DBP deficiency can accumulate VLCFA, phytanic acid, pristanic acid, and bile acid precursors (121; 74; 107; 29). The amount of phytanic acid (and, thus, pristanic acid) that accumulates depends on dietary intake. Dietary phytanic acid primarily comes from the ingestion of fats from ruminant animals (eg, beef, cow’s milk) and certain surface fish (122; 68). As such, phytanic and pristanic accumulation is typically not observed in infants. Defects in both VLCFA and pristanic acid degradation can be measured in cultured skin fibroblasts from patients with DBP deficiency (121; 97). Conversion of the 27-carbon precursor THCA to the 24-carbon primary bile acid, cholic acid, is also defective in DBP deficiency, resulting in THCA accumulation in plasma (97). Both the side chains of THCA (and DHCA) and pristanic acid are 2-methyl carboxylic acids, and their degradation proceeds by the same pathway. As bile acid synthesis takes place only in liver, skin fibroblasts cannot be used to assess the bile acid synthesis defect.
Because they contain asymmetric carbon centers, stereoisomers of phytanic acid, pristanic acid, and bile acid precursors are found in nature. Both 2R- and 2S-stereoisomers of pristanic acid are produced from the alpha-oxidation of phytanic acid, but only the 2S-isomer is a substrate for branched acyl-CoA oxidase (36). Alpha-methyl-acyl-CoA (AMACR) interconverts the two stereoisomers, thereby functioning as an auxiliary beta-oxidation enzyme. In pristanic acid degradation, the methyl groups on carbons 6 and 10 are also in the R-configuration. As pristanic acid is progressively shortened via several cycles of beta-oxidation, AMACR is again required to permit further degradation. The bile acid precursors THCA and DHCA also exist as 25R and 25S stereoisomers (because the carbon numbering system differs between fatty acids and steroids, the 2-position of pristanic acid and the 25-position of THCA/DHCA are both alpha to the carboxyl carbon). As with pristanic acid, only the S-stereoisomer can be processed by beta-oxidation into the mature bile acids. Thus, AMACR facilitates the utilization of the R-isomer. Elevations in the plasma levels of both pristanic acid and bile acid precursors are consistently found in people with AMACR deficiency. Because pristanic acid is solely derived from dietary phytanic acid, plasma levels may reflect the patient’s consumption of ruminant meats, dairy products, and certain fish.
To date, no patients with defects in any of the acyl-CoA synthetases or EHHADH have been reported. As noted above, the one report of a patient with peroxisomal thiolase (ACAA1) deficiency was incorrect.
Peroxisome morphology. In most patients with early-onset ACOX1 or early-onset DBP deficiency, and in the one patient with ABCD3 deficiency, the morphology of peroxisomes in skin fibroblasts are unusual. When examined by indirect immunofluorescence using an antibody to a peroxisomal membrane or matrix protein, these cells contain a significantly reduced number of peroxisomes of larger than usual size (79; 13). Although the reason for this change is unknown, the morphologic appearance of peroxisomes can be valuable in prenatal and antenatal diagnosis of at least the more severe forms of disease. This has not proven to be the case for more recently appreciated milder forms of disease.
Pathogenesis. It is likely that one or more compounds that accumulate in the tissues of patients with single enzyme deficiencies of peroxisomal beta-oxidation contribute to the pathologic features of these disorders. Nevertheless, correlations between specific metabolites and specific pathologic processes are lacking. For example, patients with early-onset ACOX1 or early-onset DBP deficiency accumulate VLCFA, as do patients with X-linked adrenoleukodystrophy. The development of the fetus and especially the nervous system is not affected in adrenoleukodystrophy but is disturbed in early-onset ACOX1 and early-onset DBP deficiencies. Symptoms of adrenoleukodystrophy develop later in life and are likely due to a combination of accumulation of VLCFA and secondary responses, such as an immunologically mediated destruction of myelin (81). There is evidence for increased oxidative stress in skin fibroblasts from patients with DBP deficiency (33). In ACOX1-deficient mouse oligodendrocytes, increased production of both reactive oxygen species and reactive nitrogen species was found; these increases were further potentiated by increased VLCFA levels (06). To date, the mechanistic relationship between oxidative stress, inflammation, and pathogenesis remains to be established.
On neuropathological examination, several abnormalities are found in patients with or who are presumed to have early-onset ACOX1 or early-onset DBP deficiency. Generalized or partial cerebral atrophy (72; 108), hypomyelination and demyelination (39; 72; 120), and presence of foamy macrophages have been reported (72; 120). Of particular interest is the presence of microgyria, and focal areas of cortical or cerebellar heterotopias indicating defective neuronal migration (39; 72; 120). Developmental defects such as disturbed neuronal migration might explain some of the clinical deficits associated with these disorders (28). Seizures are often an early neurologic symptom in neuronal migration defects (104). Neuronal heterotopias in other conditions, such as neurofibromatosis, myotonic dystrophy, and other muscular dystrophies, have been postulated to be among factors that result in intellectual disabilities (104).
Patients with DBP deficiency who survive beyond early childhood have progressive neurologic deficits, including ataxia and cerebellar atrophy despite normal or only mildly elevated levels VLCFA and other potentially toxic metabolites (78; 65; 58; 57). Thus, additional properties of HSD17B4 function may contribute to the pathophysiology. For example, mice with neuron-specific HSD17B4 deletion developed progressive motor disabilities and ataxia that correlated with loss of Purkinje cells and cerebellar atrophy, whereas mice with oligodendrocyte-specific knockout did not (102). Similar to observations in patients, cerebellar degeneration in mice did not correlate with VLCFA levels or other substrates of peroxisomal beta-oxidation.
The role of elevated levels of bile acid intermediates in the pathogenic neuronal development, especially the occurrence of neuronal migration defects, has been discussed. Disturbances of neuronal migration have been reported in patients with early-onset DBP deficiency (39; 19; 72; 120; 111) and early-onset ACOX1 deficiency (83; 28). Abnormal bile acid intermediates were also present in the same patients with early-onset DBP deficiency. In contrast, patients with ACOX1 deficiency have normal bile acids. It has been speculated that the mild ataxia and cognitive impairment seen in the single patient with ACOX2 deficiency resulted from increased levels of toxic bile acid synthesis intermediates (103). Somewhat surprisingly, there were no neurologic deficits reported in the single patient with ABCD3 deficiency, despite similar elevations of plasma bile acid precursor levels (34).
Adrenal atrophy is frequently reported in X-linked adrenoleukodystrophy and Zellweger spectrum disorder, which are caused, at least in part, by a deficiency in peroxisomal beta-oxidation (39; 19; 72; 120; 110; 10). Until recently (14), adrenal dysfunction has not been reported in patients with DBP deficiency. Still, adrenal cortex atrophy was reportedly found in five of 12 patients with DBP deficiency (29), which supports a prior report of small adrenal glands in another patient (120). In 2020, there was a report of primary adrenal insufficiency in two siblings with early-onset DBP deficiency (14). The authors conclude that patients with DBP deficiency should be prospectively screened for primary adrenal insufficiency and treated accordingly. There are no reports of adrenal insufficiency in early-onset ACOX1 deficiency, but this may be due to the limited numbers of patients reported to date.
Renal cysts are frequently found in severe cases of Zellweger spectrum disorder (90). In early reports, renal cortical cysts were found in two patients with early-onset DBP deficiency (39; 19). In another patient with early-onset DBP deficiency, small glomerular cysts were detected on autopsy (120). In a subsequent report, renal cysts were present in four of 12 patients with DBP deficiency (29). To date, functional impairments of kidney functions were not reported to result from such changes, and renal cysts have not been reported in patients with ACOX1 deficiency.
Insights into the pathomechanisms of ACOX2, ABCD3, ACBD5, ACOX2, AMACR, and SCP2 deficiencies are limited by the numbers of patients reported to date. For example, given that a single patient with SCP2 deficiency has been reported, its pathogenesis is poorly understood. It has been hypothesized that elevated pristanic acid levels may be responsible for the pathogenesis of AMACR deficiency and that a low phytanic acid diet may be beneficial for patients. Nevertheless, plasma exchange to acutely lower plasma pristanate levels in one patient with rapidly deteriorating neurologic status produced no clinical response (18). It has been proposed that the toxic effects of BCFAs seen in patients with AMACR deficiency may be the result of abnormal cellular signaling involving intracellular calcium levels (54). Bile acid biosynthesis defects have been attributed to the cholestasis and coagulopathy due to fat-soluble vitamin malabsorption found in patients with AMACR deficiency and, by extension, digestive abnormalities in people with ACOX2 deficiency (31). For AMACR deficiency, the adult-onset neurologic symptoms have been attributed to pristanic acid accumulation (not observed in ACOX2 deficiency), but a role for toxic levels of bile acid intermediates cannot be excluded (31).
The absolute incidence of single enzyme defects of peroxisomal beta-oxidation is unknown. As noted earlier, the clinical presentations of early-onset ACOX1 and early-onset DBP deficiencies is similar to that seen in severe cases of Zellweger spectrum disorder. In an early study involving several hundred patients, an estimated 18% of those originally diagnosed as having a peroxisome biogenesis disorder are ultimately found to have DBP deficiency (70). The incidence of peroxisome biogenesis disorders is estimated to be about 1:80,000 live births (101). As such, a crude estimate puts the incidence of DBP deficiency at about 1:440,000 live births. In a series examining 26 patients suspected of having ACOX1 or DBP deficiency, four were found to have ACOX1 deficiency, and the rest were found to have DBP deficiency (121). Fewer than 20 cases of AMACR deficiency, one case of SCPx deficiency, two cases of ACOX2 deficiency, five cases of ACBD5 deficiency, and one case of ABCD3 deficiency have been reported in the literature.
Due to the genetic nature of these disorders, the only measure of prevention is genetic counseling.
ACOX1 and DBP deficiencies. Using chorionic villus sampling or amniocentesis, cells can be obtained and the activity of the peroxisomal beta-oxidation pathway can be measured in vitro using a radiolabeled VLCFA as substrate (88; 44). The amount of VLCFA in cultured cells can also be quantitated to establish the diagnosis (69). Molecular analysis of genomic DNA for prenatal diagnosis of DBP deficiency has been reported (77). In addition, indirect immunofluorescence analysis using an antibody to a peroxisome matrix protein (eg, catalase) or a peroxisomal membrane protein (eg, PMP70) can establish whether the morphologic appearance of peroxisomes is consistent with a diagnosis of severe ACOX1 or DBP deficiency. Due to the relatively low general incidence of single enzyme defects of peroxisomal beta-oxidation discussed herein, prenatal diagnosis is usually offered only to parents with a previous affected child. If the diagnosis in the sibling was not established biochemically, prenatal testing might also be offered if the clinical presentation of the sibling was consistent with a peroxisomal defect. The tests performed to rule out a peroxisomal single enzyme deficiency will also detect patients with peroxisome biogenesis disorders. It should also be noted that in multiple published reports, the patients were born from a consanguineous union (39; 19; 79; 08; 108; 59; 27; 34; 32; 31; 12; 43; 103; 22; 05; 53; 09).
Other single enzyme defects of peroxisomal beta-oxidation. Prenatal diagnosis for these disorders has not yet been described.
Early-onset ACOX1 or DBP deficiencies. In the neonatal period, especially if dysmorphic features or early-onset seizures are absent, hypotonia will be the presenting symptom.
In the absence of an abnormal obstetrical history or any acute perinatal events (such as complicated or prolonged labor, perinatal depression with possible ischemia, acidosis, and hypoxia) a genetic or neurologic cause of the hypotonia appears more likely (02; 87). Gestational age of a hypotonic newborn should be taken into consideration because premature infants are usually somewhat, but not profoundly, hypotonic. Diseases of the motor unit (including spinal muscular atrophy, transient neonatal or congenital myasthenia gravis, myotonic dystrophy, and other congenital muscular dystrophies and myopathies) should be differentiated from disorders of the central nervous system associated with hypotonia (104; 02; 87). In addition to hypoxic ischemic events, central causes of hypotonia include other metabolic encephalopathies (eg, hyperbilirubinemia and hyperammonemia), infectious encephalitis (bacterial or viral, especially herpes simplex infection), endocrinopathy (hypothyroidism), or other genetic disorders, such as those involving amino acid or fatty acid metabolism, gangliosidosis, mucopolysaccharidosis, chromosomal abnormalities (eg, trisomy 21), and Prader-Willi syndrome. Congenital malformations of the central nervous system, such as neural tube defects, holoprosencephaly, Chiari malformation, Dandy-Walker syndrome, and neuronal migration defects, including neuronal heterotopias and lissencephaly, should be excluded by physical exam or imaging studies (116; 104).
Neonatal seizures provide evidence of CNS involvement. Seizures can also be the sequelae of a perinatal or postnatal insult to the central nervous system. Other possible causes of convulsions, such as hypoxic-ischemic encephalopathy, intracranial hemorrhage, hypoglycemia, hypocalcemia, intracranial infection, or drug withdrawal should be excluded (104). Signs that the hypotonia has already been present during fetal life, such as contractures or positional malformations of the extremities as well as decreased fetal movements by maternal report or ultrasound examination, decrease the likelihood that the symptoms are due to acute peri- or postnatal events.
The diagnosis of ACOX1 and DBP deficiencies or other single enzyme defects of peroxisomal beta-oxidation can only be established by biochemical or sequence variant analyses. Due to their similarities, it is not always possible to differentiate among ACOX1 and DBP deficiencies and Zellweger spectrum disorder. It has been reported that the patients with early-onset ACOX1 deficiency can develop more gross and fine motor skills than those with early-onset DBP, which presents similarly to Zellweger spectrum disorder (28; 90). Nevertheless, Zellweger spectrum disorder shows a wide range of phenotypic expression, including patients with atypical and less severe presentations (52; 11; 90). In six cases in which brain MRI and clinical presentation was suggestive of an early-onset peroxisomal disorder, but without clear-cut biochemical abnormalities characteristic of these diseases, variant analysis revealed that three had DBP deficiency and three had Zellweger spectrum disorder (96). The authors concluded that biochemical studies of skin fibroblasts should be performed if MRI is suggestive of a peroxisomal disorder, but blood tests of peroxisomal metabolites levels (eg, VLCFAs) are negative or equivocal.
Later-onset ACOX1 or DBP deficiency. As more adults with milder adult-onset forms of ACOX1 and DBP deficiency are identified via exome sequencing, a clearer clinical picture should emerge. Milder adult-onset DBP deficiency should be considered in the differential diagnosis of progressive gait ataxia.
Other single enzyme defects of peroxisomal beta-oxidation. Adult Refsum disease and X-linked adrenomyeloneuropathy should be considered in adults suspected of AMACR deficiency. The latter should be considered in women as well as men, as neurologic symptoms often develop in heterozygous females in later life (27). Because the biochemical abnormalities in the two disorders are similar (See Table 3), the patient with SCP2 deficiency was initially thought to have AMACR deficiency (35). A finding of normal AMACR enzyme activity in cultured skin fibroblasts from the patient prompted further investigation, which established lack of SCP2 enzyme activity and a deleterious variant in the SCP2 gene (35). ABCD3, ACBD5, and ACOX2 deficiencies are all ultrarare. Expected trends in peroxisome metabolite levels in blood specimens and cultured patient fibroblasts are noted in Table 3. Some of the clinical manifestations of ABCD3, ACOX2, AMACR, DBP, and SCP2 deficiencies result, at least in part, from impaired bile acid metabolism. Finally, white matter involvement has been observed in all patients with ACBD5 deficiency reported to date.
For patients for whom a single enzyme defect of peroxisomal beta-oxidation is considered in the differential diagnosis, the initial evaluation should include a detailed history with emphasis on genetic, family, and obstetrical history, as well as a thorough physical and neurologic examination. In some cases, a neurologic examination of the parents might be indicated. Laboratory investigation should include glucose and calcium levels as well as other electrolytes, hematologic evaluation to detect signs of infection, cultures of blood and, in some cases, cerebrospinal fluid and drug screening, if indicated. Reviews of fetal heart rate tracings, prenatally obtained scalp pH, Apgar scores, cord blood pH, and an early blood gas will help to exclude perinatal distress as contributing factors to the infant's symptoms. An EEG, or if possible, a video EEG, should be performed if seizures are present or suspected. Imaging studies such as head ultrasound, computed tomography, and magnetic resonance imaging are indicated to assess for the presence of cerebral hemorrhage, structural abnormalities, or hydrocephalus. In most patients with early-onset DBP or ACOX1 deficiencies, imaging studies were normal shortly after birth (79; 120; 59). Some mild abnormalities were observed in other patients, including mild frontal atrophy (72), decreased density of the white matter (39), and minimal white matter hypodensities (79). Later in life, white matter abnormalities may be more pronounced (79).
Sequencing is currently a primary tool to establish or confirm a suspected single enzyme defect of peroxisomal beta-oxidation. Although clinical genetic analyses are widely available, most of the biochemical analyses required to establish an accurate diagnosis of peroxisomal diseases are specialized and are only available at a few centers worldwide. The initial test to establish the diagnosis of a peroxisomal defect should be quantitation of plasma VLCFA levels (71). If this test is requested early in the evaluation, invasive procedures to establish a diagnosis, such as muscle or even brain biopsy, can be avoided. Blood VLCFA levels are strikingly elevated in most patients with early-onset ACOX1 and early-onset DBP deficiencies and X-linked adrenoleukodystrophy, but not for the other single enzyme defects of peroxisomal beta-oxidation discussed herein and are not significantly altered by the timing of the blood drawing, the patient's nutritional status, anticonvulsant medications, or age. If VLCFA levels are abnormally elevated, additional studies to exclude other peroxisomal dysfunctions present in peroxisome biogenesis disorders should be performed (42). These tests include measuring plasma phytanic acid, pristanic acid, and pipecolic acid levels as well as red blood cell plasmalogen levels and bile acid intermediates. Plasma phytanic and pristanic acid levels can be elevated in AMACR, SCP2, and DBP deficiencies as well as in peroxisome biogenesis disorders. Because phytanic acid (and, therefore, its metabolite, pristanic acids) only comes from the diet, the degree of elevation is dependent on the age and diet composition of the patient. Nevertheless, in a series of 126 patients with DBP deficiency studied at the Academic Medical Center in Amsterdam, three had normal plasma VLCFA, phytanic acid, and pristanic acid levels, although skin fibroblast VLCFA levels were elevated (29). The older sibling with Perrault syndrome described above also had normal serum VLCFA levels when measured at 23 years of age (78). A similar phenomenon was also reported for one patient with ACOX1 deficiency (83). Pipecolic acid, which accumulates in peroxisomal biogenesis disorders due to lack of peroxisomal L-pipecolate oxidase, can be mildly elevated in early-onset DBP deficiency for reasons that are unclear (39; 120; 08).
The analysis of plasma, urine, or bile will usually reveal the presence of abnormal bile acid intermediates (THCA, DHCA, or other metabolites) in patients with DBP, SCP2, AMACR, ACOX2, and ABCD3 deficiencies (39; 120; 111; 109; 36; 29; 35; 34; 103) but not in patients with ACOX1 deficiency. However, about one-fourth of patients with DBP deficiency show no accumulation of plasma THCA or DHCA (29). Although both 25R- and 25S-stereoisomers accumulate in DBP deficiency and peroxisomal biogenesis disorders, only the 25R-isomer accumulates in AMACR deficiency.
The measurement of urinary acyl-carnitines by multiple reaction monitoring (MRM) electrospray ionization tandem mass spectrometry (ESI-MS/MS) has emerged as a promising method for diagnosis of peroxisome biogenesis disorders and DBP deficiency (24). In a study of seven patients with peroxisome biogenesis disorders and two patients with DBP deficiency, significantly elevated levels of 14-. 16-, and 18-carbon dicarboxylylcarnitines and 22-, 24-, and 26-carbon monocarboxylylcarnitines were detected in all but one sample. The level of 16-carbon dicarboxylylcarnitine in the urine of only one patient with DBP deficiency was markedly elevated. Nonetheless, the authors conclude that MRM ESI-MS/MS is a rapid, reliable, and sensitive diagnostic method that discriminates patients with these disorders from controls.
If a diagnosis of peroxisomal disorder is suspected, it is valuable to obtain a skin biopsy to establish cultured fibroblasts. The overall rate of peroxisomal beta-oxidation of a VLCFA (eg, lignoceric acid; C24:0) or pristanic acid can be measured in fibroblasts using [14C]-labeled substrates (88; 44; 27; 100). Wanders and coworkers developed an HPLC assay using varanoyl-CoA, a beta-oxidation intermediate between THCA-CoA and choloyl-CoA in bile acid synthesis, as substrate to diagnose DBP deficiency (105). Metabolites formed when this substrate is incubated with fibroblasts from patients suspected of having DBP deficiency indicate whether there is a deficiency of the enzymes’ enoyl-CoA hydratase activity, its 3-hydroxyacyl-CoA dehydrogenase activity, or both.
Immunoassays that detect proteins relevant to single enzyme defects of peroxisomal beta-oxidation in liver biopsy samples, cultured fibroblasts, and postmortem tissues from patients can be useful for diagnosis (44; 98; 49; 100). In more severe cases, the protein of interest may be undetectable by such methods (85; 79; 120); however, in some patients the variant protein of interest is often present but catalytically inactive. In such cases, immunoassays are less informative. In early studies, complementation techniques involving the fusion of cells from different patients were used to establish the diagnoses of ACOX1 and DBP deficiencies (63; 92; 121). Although powerful, these classic methods have been supplanted by modern genetic analyses.
ACOX1 and DBP deficiencies. There are no curative treatments for early- or later-onset ACOX1 and DBP deficiencies. After establishing a diagnosis, supportive measures should be initiated to assist the caregivers in coping with the nature and prognosis of the patient’s condition, which can range in severity. It is important to emphasize the variable severity of these disorders, especially in cases where the patient has a mild form of disease compatible with a prolonged lifespan. Guardians and caregivers should be informed of the genetic nature of the disease. Providing care to a child with a genetic disorder can require enormous adjustments in family life. Although the family or guardians might be concentrating on their affected child's diagnosis, care, and immediate needs, they should receive genetic counseling to understand the risks for consequent pregnancies and their options, including prenatal diagnosis.
Feeding difficulties are frequently reported in patients with early-onset ACOX1 and DBP deficiencies. Although some patients have sufficient suck and swallow coordination to be bottle fed, others require gavage tube feedings (39; 109). Placement of a gastrostomy tube might be indicated (120). Failure to thrive can complicate the course in these patients (39; 108). Assistance should be provided for the general care of the infant and specifically for feeding. Appropriate positioning is essential to prevent secondary deformities. Medical management should focus on seizure control and prevention or early detection of possible complications.
The characteristic severe hypotonia will require the caregiver to provide most of the support when holding the infant. Foam contoured seats, Velcro straps, wedges, or rolls might be necessary to maintain good position in a baby seat. With appropriate support of head, trunk and extremities, secondary complications such as torticollis, scoliosis, contractures of the hips in external rotation and abduction, knee contractures, ankle and feet deformities might be avoided; physical therapy is an additional measure of prevention. In one patient (120) a femur fracture occurred at age 3.5 months without a history of trauma. On skeletal survey, generalized osteopenia was present. Delayed bone maturation has been reported in several cases (19; 120).
Despite generalized hypotonia, respiratory drive and chest excursions seem sufficient in these patients to maintain adequate ventilation. No patient required long-term mechanical ventilation after birth. Chest physical therapy and suctioning might be required in some patients to prevent secondary complications.
Seizure control seems to be extremely difficult to achieve in patients with early-onset ACOX1 and DBP deficiencies. Seizures were controlled with phenobarbital treatment in only one reported presumed case (08). Frequent convulsions can occur in early-onset DBP deficiency despite multidrug therapy, including phenobarbital, phenytoin, and, in older patients, valproic acid and primidone (72). Due to possible impairment of adrenal function, corticotropin treatment may not be effective. One patient with an early-onset DBP deficiency received prednisone instead but with only temporary improvement (72). A ketogenic diet has not been used and should only be considered if VLCFA levels are followed because elevation of these fatty acids has been observed in some patients receiving a ketogenic diet for the treatment of uncontrollable seizures (93).
Liver and adrenal functions should be followed in patients with early-onset ACOX1 and early-onset DBP deficiencies (Table 1). Individuals with early-onset DPB deficiency are at risk of developing adrenal dysfunction (14). It is also possible that some patients with milder forms of early-onset ACOX1 and early-onset DBP deficiencies may also be at risk for adrenal and liver dysfunction.
As previously noted, malformations have been associated with early-onset DBP deficiency, including ventricular septal defects (39; 72). The clinical course of one patient with DBP deficiency was complicated by a hemodynamically significant ventricular septal defect leading to intermittent cardiac failure and required treatment with diuretics and digoxin (39). No other cardiac malformations have been reported in patients with single enzyme defects of peroxisomal beta-oxidation.
A hematopoietic stem cell transplant was performed in one patient with ACOX1 deficiency at 3 years of age (115). MRI changes were first noted at 23 months and showed only mild progression at the time of transplant. Despite full engraftment, he continued to decline neurologically and died of pneumonia at 6.75 years of age. Although the transplant did not halt progression of his disease, neuroimaging studies showed evidence of slowing, with less brain inflammation, cortical atrophy, and neuronal loss than observed in his affected older brother, who was not transplanted.
SCP2, ACOX2, and ABCD3 deficiencies. The treatment of the single reported patient with SCP2 deficiency was not described (35). One ACOX2 patient was treated unsuccessfully with ursodeoxycholic acid; nonetheless, the authors speculated that this disorder could potentially be treated with bile acid therapy (103). The rationale for this approach is that mature bile acids would block early steps in bile acid synthesis, decreasing the levels of toxic intermediates. Although the single patient with ABCD3 deficiency died before the diagnosis was established (34), the biochemical similarities between ABCD3 and ACOX2 deficiencies could suggest that bile acid therapy may be worth discussing with appropriate colleagues.
AMACR deficiency. Pristanic acid, which is suspected but not proven to contribute to the pathology of AMACR deficiency, is derived solely from the dietary ingestion of phytanic acid. It has been suggested that a low phytanic acid diet could lower pristanate stores in patients (18; 94). Symptomatic treatment of seizures and other neurologic manifestations is indicated. The child with AMACR deficiency who presented in the neonatal period with coagulopathy and mild cholestasis was treated with cholic acid and fat-soluble vitamin supplementation and was reportedly in good health at 7.5 years of age (86). However, it should be noted that this is consistent with the natural course of disease. As noted above, the liver from this patient’s older sibling, who died at 5.5 months of age following an acute episode later ascribed to a coagulopathy, was used for transplantation. Because a posttransplant biopsy revealed evidence of acute rejection, bile-duct proliferation, and fibrosis, the recipient was treated with ursodeoxycholic acid. This child was reportedly doing well 8 years posttransplant and was still receiving ursodeoxycholic acid.
Although it is theoretically possible that women with milder cases of ACOX1 or DBP deficiencies or other single enzyme defects of peroxisomal beta-oxidation could become pregnant, it has not been discussed in the literature. In healthy women, pregnancies that give rise to infants with single enzyme defects of peroxisomal beta-oxidation are typically described as being uncomplicated. Nevertheless, severely affected infants may have had decreased fetal movements as an early sign of hypotonia. There are reports of patients with DBP deficiency (39; 72; 120; 14; 84; 16) and ACOX2 deficiency (31) being delivered by cesarean section. The indication in one case was breech position (72); it can be hypothesized that the fetal hypotonia might have contributed to the abnormal breech presentation. Labor was well tolerated in most babies; the presence of meconium indicated possible fetal distress in only two cases (59). One patient with early-onset DBP deficiency was born prematurely (at 34 weeks); the neonatal course was complicated by episodes of hypoglycemia and a hemorrhagic diathesis (19).
No specific information is available. There are no reports of airway abnormalities in patients with single enzyme defects of peroxisomal beta-oxidation. Depending on the medications used, it should be taken into consideration that some patients may have impaired liver functions.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Joseph Hacia PhD
Dr. Hacia of the University of Southern California received consulting fees and stocks from Congruence Therapeutics as a consultant and scientific co-founder.
See Profile
Femke Klouwer MD PhD
Dr. Klouwer of Amsterdam UMC has no relevant financial relationships to disclose.
See Profile
Andrea Gropman MD
Dr. Gropman of St. Jude Children's Research Hospital has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neurogenetic Disorders
May. 08, 2026
Neurogenetic Disorders
Apr. 30, 2026
Neuropharmacology & Neurotherapeutics
Apr. 23, 2026
Neurogenetic Disorders
Apr. 14, 2026
Neurogenetic Disorders
Apr. 14, 2026
Neurogenetic Disorders
Apr. 14, 2026
Neurogenetic Disorders
Apr. 14, 2026
Neurogenetic Disorders
Apr. 14, 2026