

Neuro-Oncology
Turcot syndrome
May. 27, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Sporadic schwannomas and neurofibromas are tumors that arise from the nerve sheaths of cranial nerves, nerve roots, spinal nerves, and peripheral nerves. Most schwannomas originate from the eighth cranial nerve, whereas neurofibromas more commonly arise along the spinal nerve roots. Often, these tumors can be followed clinically and radiographically. Some, however, require treatment. Maximal surgical resection is the treatment of choice for many of these tumors, but radiation therapy can be appropriate in selected cases. Systemic therapies are still under investigation for therapeutic potential. There has been recent regulatory approval for MEK inhibition for a subset of patients with neurofibromas occurring in the setting of neurofibromatosis type 1 (NF1). In this update, the authors discuss the pathophysiology, clinical presentation, treatment, and outcomes of these conditions.
|
• Schwannomas are benign (WHO grade I) slow-growing, extra-axial tumors that can arise from Schwann cells in any cranial nerve, spinal nerve root, or peripheral nerve. They can frequently arise from the eighth cranial nerve. | |
|
• Neurofibromas are usually benign (WHO grade I) peripheral nerve sheath tumors that involve Schwann cells as well as nerve fibers, fibroblasts, and cutaneous tissue. They can occur in superficial (cutaneous, subcutaneous) and deep (visceral, spinal, orbital) locations. | |
|
• When intervention is indicated, surgical resection is the treatment of choice for most schwannomas and neurofibromas. Complete resection can be curative. | |
|
• In selected cases, stereotactic radiosurgery (SRS) or fractionated radiotherapy may be effective for local control of tumor growth | |
|
• There is currently no well-defined role for systemic therapy in sporadic schwannomas and neurofibromas. However, MEK inhibition has received regulatory approval for a subset of patients with neurofibroma occurring in the setting of neurofibromatosis type 1. |
Cruveilhier is credited with the most complete initial description of both the clinical and pathologic features of a vestibular schwannoma (15). Sir Charles Balance performed the first successful surgical resection of a vestibular schwannoma in 1894. The neurosurgical community thereafter quickly developed interest for vestibular schwannoma surgery (137), and over the next 30 years, surgical techniques were refined to such a degree that operative mortality was reduced from 50% to 60% in the early 1900s to 4% by 1931 (74). Additional advances, such as the advent of antibiotics, anesthesia, and refinement in surgical techniques have further improved surgical morbidity and mortality.
Schwannomas and neurofibromas are benign (WHO grade I) tumors arising from peripheral nerve sheaths (71). Schwann cells are the neoplastic cells of origin in both schwannomas and neurofibromas, but the latter also contain intratumor nerve fibers and a large variety of additional cell types, such as fibroblasts, perineurial cells, and mast cells (122).
|
• Tumors stemming from peripheral nerve sheaths, sporadic schwannomas, and neurofibromas can originate from cranial nerves, spinal nerves, nerve roots, or peripheral nerves. Therefore, the clinical manifestations and evolution are variable, depending on the tumor location, size, and rate of growth. | |
|
• Nonsporadic schwannomas occur in the context of neurofibromatosis type 2 (NF2) related schwannomatosis and non-NF2-related schwannomatosis, whereas neurofibromas are cardinal manifestations of neurofibromatosis type 1. These hereditary conditions are discussed in other appropriate articles. |
Vestibular schwannomas. Vestibular schwannomas account for the vast majority of sporadic schwannomas. Because the Schwann cell-glial junction is variable, the tumor can arise from a lateral position near the inner ear or inside the labyrinth, to a medial position at the porus acusticus, to completely within the cerebellopontine angle (121; 39).
Patients with vestibular schwannomas present with (85):
|
• Cochlear nerve dysfunction, as manifested by unilateral sensorineural hearing loss (95%) and tinnitus (63%). Hearing loss may have been present from 1 to 5 years (mean 3.7 years) prior to diagnosis. In most cases, the loss is gradually progressive, but it can present acutely in 10% to 20% of patients (possibly due to occlusion of the internal auditory artery). Sensorineural hearing loss develops from a combination of tumor-induced interference with cochlear vascular supply and compression of the cochlear nerve within the internal auditory canal. Tinnitus is often unilateral, confined to the affected ear, low grade in intensity, and constant. | |
|
• Vestibular nerve dysfunction manifested by gait unsteadiness (61%). Due to the slow-growing nature of these tumors, acute vertigo is uncommon. Of note, intralabyrinthine schwannomas that have an intravestibular component are characterized by significantly worse vestibular function compared to schwannomas with purely cochlear involvement (144). | |
|
• Involvement of the trigeminal nerve (17%), with facial hypoesthesia, paresthesia and pain. | |
|
• Facial nerve compression (6%) leading to facial paralysis and in some cases, taste disturbance, xerophthalmia, xerostomia, or lacrimation. | |
|
• Symptoms due to tumor mass effect on the adjacent infratentorial matter, such as ataxia, hydrocephalus, cerebellar herniation, and lower cranial nerve dysfunction. |
Some patients also report subacute or chronic headache (29%). Although rare, patients can present with an acute severe headache associated with nausea and emesis from schwannomas that have hemorrhaged (58).
Trigeminal schwannomas. Trigeminal schwannomas usually arise from the trigeminal ganglion (121). Affected patients present with facial numbness (25% to 30%), pain (20% to 25%), and paresthesia (5% to 10%). Other symptoms may include headache (15%), diplopia (10%), and hearing loss/tinnitus (10%) (87). Notable clinical examination findings include ipsilateral trigeminal dermatome hypoesthesia, a weak or absent ipsilateral corneal reflex, limitations in extraocular movement, pyramidal signs by mass effect on the pons, dysmetria, and ataxia, and exophthalmos by rostral mass effect.
Other cranial nerve schwannomas. Ocular motor nerve schwannomas (III, IV, VI) are extremely rare (125; 54; 47; 48). They arise in the interpeduncular cistern more frequently than in the cavernous sinus or the orbit, and present with diplopia and headache. Patients may also have a variety of symptoms and signs localizing to the causative or adjacent structures (eg, visual loss, facial sensory loss, ataxia, proptosis).
Facial nerve schwannomas arise from the vertical segment of the nerve (within the temporal bone), or less often, near the tympanic membrane or cerebellopontine angle (121). Patients mostly report facial weakness and hearing loss (50% to 90%), tinnitus (60%), and vertigo (34%). The symptoms are slowly progressive in 80% of patients (mean 6 to 7 years), whereas in the remainder, the symptoms have an acute or fluctuating onset (106).
Lower cranial nerve schwannomas (IX, X, XI) may present with the classic jugular foramen syndrome: dysphagia, weakness and atrophy of sternocleidomastoid and trapezius muscles, hoarseness, and diminished taste (125; 07). When the tumor protrudes significantly into the posterior fossa, as is the case most often with glossopharyngeal schwannomas, patients may present with symptoms similar to those of vestibular schwannomas (125; 138).
Hypoglossal schwannomas are very uncommon and can present with tongue atrophy and weakness, vertigo, headache, and other brainstem compression symptoms.
Spinal schwannomas. Spinal schwannomas usually develop from the nerve roots, with a predilection for the dorsal sensory branches. The evolution of symptoms is slowly progressive, with a median symptom-onset to diagnosis delay of a year (14). Symptoms and signs include radicular (72%) and/or localized (59%) pain, peripheral weakness (60%), incontinence (33%), and gait difficulty (28%) (14; 109). Depending on the lesion localization, there may be myelopathic/upper motor neuron as well as lower motor neuron signs.
Peripheral nerve schwannomas. These tumors can arise on any peripheral nerve structure but are most common in the brachial plexus and flexor peripheral nerves, especially the elbow, knees, and wrists (56). Patients initially notice a painless mass, and with time, it may progress to become tender and/or cause compressive symptoms, such as hypoesthesia or paresthesia. On examination, patients typically have a tender mass that can be displaced from side to side, but not longitudinally. Percussion of the mass can often induce painful paresthesia in the distribution of the nerve. In most cases, there are no sensory and motor deficits in the distribution of the affected nerve. Most peripheral nerve schwannomas progress very slowly, but a minority (13%) of patients harbor fast-growing lesions (29). There is no correlation between initial tumor size, age at diagnosis, and tumor growth rate.
Neurofibromas. Similar to peripheral nerve schwannomas, sporadic neurofibromas tend to present as painless masses. Recent neurofibroma classifications have divided these tumors into superficial (cutaneous or subcutaneous) and deep (visceral, spinal or orbital) categories (46).
Arising from a peripheral nerve lying within the dermis or epidermis, a cutaneous superficial neurofibroma presents as a soft gelatinous sessile or pedunculated (“wart-like”), nontender, mobile nodule. There can be an overlying violaceous discoloration. These tumors do not carry a risk of malignant transformation, but they often cause esthetic issues or functional problems mandating surgical intervention.
Plexiform neurofibromas are similar to their cutaneous counterparts, but they tend to involve multiple nerve fascicles or a nerve plexus. When plexiform neurofibromas occur subcutaneously, they are palpable and can either be nodular (rubbery, firm, linear, and beaded) or diffuse (elastic and poorly defined, as a “bag-of-worms”). Unlike schwannomas, these tumors can occasionally cause neurologic deficits. When plexiform neurofibromas are located internally, they are nonpalpable masses involving the cranial cavity, orbit, cranial nerves, spine, or viscera (67). In these cases, clinical presentation and evolution are similar to that of schwannomas arising in the same location (112; 116). The lifetime risk for plexiform neurofibroma malignant transformation is estimated to be 10% (01).
Malignant peripheral nerve sheath tumors. Malignant peripheral nerve sheath tumors (MPNST) most commonly occur in patients with neurofibromatosis type 1, but they can arise in patients with sporadic neurofibromas, particularly nodular and plexiform neurofibromas. Malignant peripheral nerve sheath tumors present similarly to neurofibromas, but symptoms tend to evolve more rapidly and more commonly include motor and sensory deficits attributed to parent structure mononeuropathy or plexopathy, as well as progressive pain.
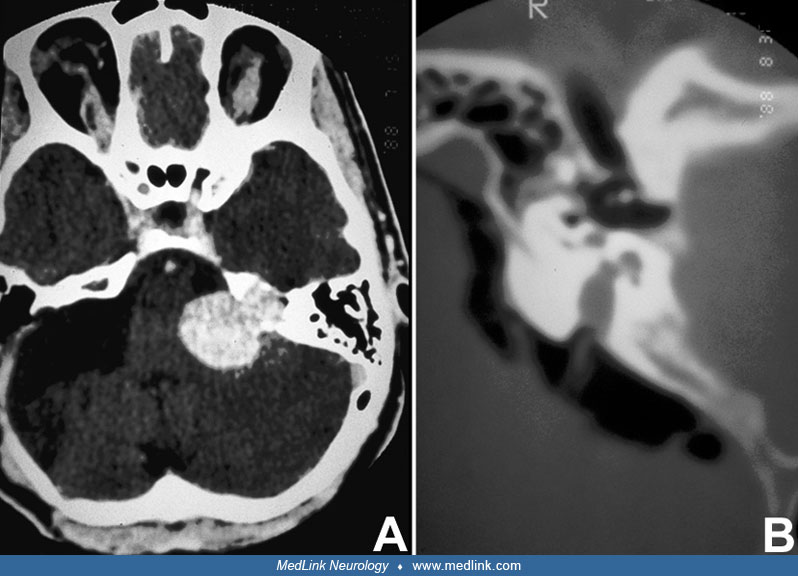
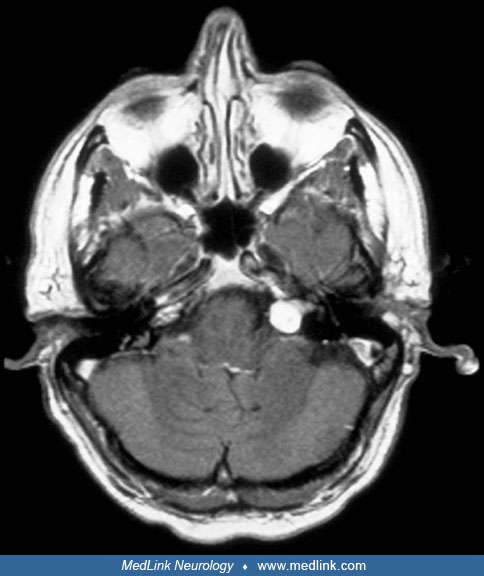
A 56-year-old male with an unremarkable past medical history progressively developed dizziness, tinnitus, and left-sided hearing loss over the span of 12 months. The initial neurologic examination was unremarkable. Audiometry confirmed mild left neurosensorial hearing loss. A left vestibular schwannoma was suspected following discovery on MRI of a 1.0 x 1.5 cm enhancing mass in the left cerebellopontine angle, with a tail that entered the internal auditory canal. The patient was subjected to a suboccipital surgical approach. The cochlear division of cranial nerve VIII was draped over the mass, which was attached to the vestibular division of VIII. After the mass was dissected away from the vestibular division, the tumor was removed piecemeal, with preservation of the facial nerve. The patient did well after surgery, with partial preservation of left-sided hearing and only mild left facial weakness, which improved after several months. Follow-up MRIs did not disclose tumor recurrence.
Sporadic schwannomas and neurofibromas arise from Schwann cells that provide myelinization of cranial nerves, spinal nerve roots, or peripheral nerves. Although the initial genesis of cellular neoplastic transformation is unknown in most cases, various contributory cytogenetic, chromosomal, and molecular biological events are discussed below.
Pathophysiology. In the most recent WHO Classification of Tumors of the Central Nervous System, Fifth Edition, schwannomas and neurofibromas are classified as cranial and paraspinal nerve tumors (71). Schwannomas are composed of a homogeneous mass of transformed Schwann cells in a collagenous background. Neurofibromas contain numerous transformed Schwann cells but also include a mixture of transformed perineurial cells and fibroblasts.
Macroscopic features. Schwannomas always arise at the transition zone between the central glial nerve sheath and the peripheral Schwann cell nerve sheath. Rarely, schwannomas can occur within the substance of the brain or spinal cord (120). There is a predilection for schwannomas to affect sensory nerves, although motor nerves can be involved as well. On gross pathologic inspection, schwannomas appear as discrete, rounded, firm, encapsulated masses of a semitranslucent or milky white color arising from a nerve fascicle. The tumors may have variable amounts of cyst formation, yellowish areas of xanthomatous changes, and hemorrhage.
Sporadic neurofibromas typically arise from small cutaneous terminal nerves, but they can also develop in large peripheral nerves, spinal nerves, or spinal nerve roots (108). On gross pathologic examination, neurofibromas are soft, well-circumscribed, pedunculated, and unencapsulated gelatinous masses of a whitish or opalescent color. Regions of cyst formation, xanthomatous changes, and hemorrhage are not seen as commonly as they are with schwannomas. During initial phases of growth, the tumor infiltrates the parent nerve, causing a localized, fusiform swelling. As the tumor enlarges, the parent nerve and those nerves around it may develop gross alterations of shape and can become encased within the mass.
Microscopic features. On histological examination, schwannomas are classically composed of a heterogeneous, biphasic architecture that contains two distinct regions, Antoni A (dense) and Antoni B (loose) (125). Nuclear palisading (Verocay bodies) is also present. In most tumors, the Antoni A regions predominate and are organized into dense, compact rows or arrays of elongated, spindle-shaped cells that have hyperchromatic, rod-shaped nuclei and eosinophilic cytoplasm. The tissue in Antoni B regions is characterized by large numbers of organelles (eg, mitochondria, lysosomes, osmiophilic bodies) and vacuoles. Immunohistochemical typing of schwannomas demonstrates strong reactivity to S-100 protein (in contrast to meningiomas), as well as Leu-7 and myelin basic protein. Therefore, cytoplasmic S-100 protein is often used to identify nerve sheath tumors (74).
Biological features. In general, studies measuring the biological activity of schwannomas are consistent with benign neoplastic processes, demonstrating small growth fractions and limited aggressive biological potential (17; 10). There is growing interest for the oncogenic potential of the inflammatory microenvironment in vestibular schwannomas (41).
The vestibular schwannoma microenvironment is determined by an interplay between stromal and immune cells that produce and remodel extracellular matrix, vascular networks, and promote tumor growth.
Angiogenesis seems to have an impact on tumor microenvironment, by influencing tumor growth index and prognosis. Even though vestibular schwannoma are generally slow-growing tumors, an increased vascularization has been found with delicate blood vessels on the adjacent vestibulocochlear nerve, susceptible to mechanical stress. This suggests that one of the most plausible explanations for hearing loss in vestibular schwannoma is edema of the cochlear nerve due to circulatory disturbances and postoperative hearing loss is likely due to blood vessel surgical injury (12).
Marioni and colleagues also investigated neoangiogenesis in sporadic vestibular schwannoma using CD105 staining and found that CD105 can serve as a useful marker to identify proliferating endothelium (83). It has finally been reported that vestibular schwannoma tumors stain positive for erythropoietin (EPO) and that tumors with higher EPO levels tend to be larger (20).
Immune system functions are also involved in tumor tissue formation. A positive correlation has been shown between tumor tissue inflammation and duration of clinical symptoms (65). When it comes to the highly studied tumor-associated macrophages (TAMs), M2-type macrophages which are known to have tumor-promoting features appeared to be highly expressed in vestibular schwannomas. Macrophage colony stimulating factor (M-CSF), which controls macrophage recruitment, proliferation, and transition towards a pro-tumoral M2-like phenotype, is highly expressed in fast-growing vestibular schwannomas, as well as cystic tumors (18) and can, therefore, be therapeutically targeted. Rapidly progressing vestibular schwannomas are also rich in CD68+ macrophages, CD4+ and CD8+ T lymphocytes, and CD20+ B cells (12).
Genetic features. Biallelic inactivation of the neurofibromin 2 gene (NF2, located on the q12 band on the long arm of chromosome 22), which is known to produce a tumor suppressor protein named merlin (also referred to as schwannomin), is found in most cases of sporadic vestibular schwannomas (148; 35). The most common mutations are small deletions that result in frameshifts (143). Protein expression studies have not revealed the presence of truncated or abnormally sized merlin products in NF2 muted cells, suggesting that mutant merlin proteins are unstable and may undergo rapid degradation (43). Epigenetic alterations of the NF2 gene may also play a role in schwannoma tumorigenesis (37). Overexpression of CPI-17, which can drive Ras activity and promote tumorigenic transformation via merlin inhibition, has been documented in sporadic vestibular schwannomas, suggesting this protein has a role in schwannoma oncogenesis (148). Other genes that are found to be mutated in sporadic vestibular schwannoma include SYNE1, IRS2, APC, CIC, SDHC, BRAF, NUMA1, EXT2, HRAS, BCL11B, MAGI1, RNF123, NLRP1, ASXL1, ADAMTS20, TAF1L, XPC, DDB2, and ETS1 (35).
Analysis into the angiogenic mechanisms underlying schwannoma growth has focused on the expression of vascular endothelial growth factor (VEGF) and its receptor (VEGFR-1) (11; 21). The concentrations of both VEGF and VEGFR-1 seem to correlate with tumor growth rate, but not with tumor size or symptom duration. Furthermore, VEGF expression appears to be greater in malignant peripheral nerve sheath tumor than WHO grade 1 schwannomas and neurofibromas (139). VEGF expression is also significantly higher in recurrent sporadic schwannomas than inaugural tumors, suggesting the protein may have a role in prognosis.
A study suggested that mechanisms including lipid metabolism, as well as Hippo and FOxO pathway dysregulation, may play a role in large vestibular schwannoma growth. They identified five target genes (SCD, LMNB2, TMEM43, JARID2, and CCND1 9) that were enriched and nine miRNAs that were abnormally expressed (miR-7, miR-142 miR-155, miR-342, miR-1269, miR-4664, and miR-6503 were upregulated, whereas miR-204 was down-regulated) in large compared to small intracanalicular vestibular schwannomas (69).
Furthermore, it was shown that nearly a third of sporadic schwannomas are characterized by recurrent in-frame insertion/deletion mutations of SOX10 while lacking changes in known nerve sheath tumor genes. This gene encodes for a transcription factor that controls Schwann cell myelination and differentiation. SOX10 mutation appears to be significantly enriched in schwannomas arising from nonvestibular cranial nerves (eg facial, trigeminal, vagus) and is absent in NF2-related vestibular nerve schwannomas. Functional studies have revealed that these SOX10 indel mutations have preserved DNA binding capacity but disrupted transactivation of myelination and glial differentiation programs (146).
The molecular basis underlying sporadic neurofibromas remains largely unexplored because virtually all tumors analyzed to date have been obtained from patients with neurofibromatosis type 1.
|
• Vestibular schwannomas account for the majority of sporadic schwannomas. | |
|
• The vast part of cases (90%) are unilateral and occur outside of the context of neurofibromatosis type 2. The mean age at diagnosis is 50 years old, with no predominance in gender or lateralization. | |
|
• Radiation exposure, particularly in childhood, is a well-recognized risk factor for the development of vestibular schwannomas. There are conflicting data on the association between vestibular schwannoma incidence and prior phone use. | |
|
• Neurofibromas usually occur in young patients, with a mean age at diagnosis of 20 to 30 years. The grand majority of cases (90%) are solitary and not associated with neurofibromatosis type 1. |
Schwannoma epidemiology. Schwannomas account for 6% to 8% of all primary central nervous system tumors, and most cases occur in adulthood (103; 64). The vast majority are sporadic and not associated with neurofibromatosis type 2-related schwannomatosis or non-NF2-related schwannomatosis. Overall, sporadic schwannomas have a peak incidence between 20 and 50 years, without gender or race predominance (96).
Vestibular schwannomas. The majority of cranial nerve VIII schwannomas arise from the vestibular branch of the vestibulocochlear nerve. The incidence of vestibular schwannoma is estimated to be of 0.8 to 1.09 cases per 100,000 population (103; 32; 64; 61), with 2.93 cases per 100,000 in the 65 to 74-year-old age group (64). Sporadic vestibular schwannoma incidence rate has been estimated at 3.0 to 5.2 cases per 100,000 person-years, with the highest rate reported among patients greater than or equal to 70 years of age (80). Overall, the incidence appears to be increasing in recent decades, due in part to incidental diagnoses (68). However, data suggest that increased incidence cannot be solely explained by greater detection alone (82). The mean age at diagnosis is approximately 50 years (103), with schwannomas occurring equally among both genders. Over 90% of schwannomas are unilateral, and there is no lateral predominance (27). Bilateral schwannomas are diagnostic for neurofibromatosis type 2-related schwannomatosis (134).
Other schwannomas. Among all intracranial schwannomas, trigeminal nerve schwannomas represent 0.8% to 8% of cases, whereas facial nerve schwannomas account for 0.5% to 1.9% of cases (125). Schwannomas of the other cranial nerves are extremely rare. The incidences of spinal and peripheral nerve schwannomas are unclear, but the former represent 15% to 50% of extramedullary spinal tumors (59).
Schwannoma risk factors. Epidemiological studies have attempted to identify risk factors for the development of sporadic vestibular schwannomas.
Family history. There have been reports in the literature that support a genetic or familial association between sporadic vestibular schwannoma cases that occur in patients who do not meet criteria for neurofibromatosis type 2-related schwannomatosis (04; 26). It is possible that some of these patients may represent mosaic neurofibromatosis type 2.
Radiation exposure. There is an association between the occurrence of vestibular schwannomas and prior radiation exposure. In a cohort of 3112 patients who had undergone therapeutic irradiation as children, 43 (1.38%) later developed a vestibular schwannoma, with a mean latency of 38.3 years. The relative risk was 1.14 per Gy of exposure (113). There does not appear to be an association between low-frequency magnetic field exposure and vestibular schwannoma incidence (34).
Phone use. Early metaanalyses investigating the possible association between cell and cordless phone use and subsequent brain tumor development found an increased risk for ipsilateral brain tumor incidence (overall odds ratio [OR] 1.6-1.9) in people whose usage exceeded 10 years (42; 55). The increased risk was observed for astrocytomas and vestibular schwannomas, but not for meningiomas. In a later study involving two Danish nationwide cohorts totaling 2.9 million subjects, there was no increased risk for the development of a schwannoma among phone users (115). It is possible that recall bias may be impacting these results. Of the tumors that did develop, tumors were not larger than expected or more likely to be on the ipsilateral side of phone use. In the INTERPHONE Study Group report, which studied 1105 patients with newly diagnosed vestibular schwannomas and 2145 case-matched controls, there was no association between vestibular schwannoma incidence and the use of a cell phone (OR 0.85), even among people who had 10 years of cell phone usage or more (OR 0.76) (49). No trends were noted for subjects with increasing cumulative call time or number of calls. In conclusion, a metaanalysis concluded that current data do not support an association between cell and cordless phone use and vestibular schwannoma incidence (90).
Smoking. There are studies supporting a protective role of cigarette smoking in developing vestibular schwannoma (26).
Medications. Certain medications seem to be protective against vestibular schwannoma, such as aspirin. Pharmacologic treatment of sporadic vestibular schwannoma with aspirin has, therefore, been proposed as a treatment alternative. It is thought to slow vestibular schwannoma growth through the inhibition of cyclooxygenase-2 (26). Metformin also appears to reduce the odds of vestibular schwannoma growth (72). At this time, these are not validated therapeutic approaches.
Noise exposure. Data on noise exposure as a possible risk factor for vestibular schwannoma are conflictual, with most studies relying on self-reported measures of exposure prone to recall bias (27). In a large epidemiological study based in Sweden, occupational exposure to mercury (OR 2.9) and benzene (OR 1.8) were associated with vestibular schwannoma incidence (102).
Neurofibroma epidemiology. Neurofibromas usually occur in young patients, with a mean age at diagnosis of 20 to 30 years. The grand majority of cases (90%) are solitary and not associated with neurofibromatosis type 1 (96). The exact incidence of sporadic neurofibromas is currently unknown.
Malignant peripheral nerve sheath tumor epidemiology. As noted previously, approximately 10% of patients with plexiform neurofibromas develop malignant peripheral nerve sheath tumor (01), with sporadic cases occurring at a later age than those associated with neurofibromatosis type 1 (mean age 40 to 45 years vs. 25 to 35 years, respectively) (70). Although 20% of patients with sporadic malignant peripheral nerve sheath tumor report having suffered prior malignancy, it remains unclear if prior personal or familial malignancy is truly a risk factor for malignant peripheral nerve sheath tumor (95). Prior radiotherapy is a well-documented risk factor for acquired malignant peripheral nerve sheath tumor in both patients with and without prior history of an underlying peripheral nerve sheath tumor (30).
No measures are known to prevent the development of sporadic schwannoma or neurofibroma in healthy persons or in those at risk from associated disorders (eg, NF1, NF2-related schwannomatosis, or non-NF2-related-schwannomatosis).
The differential diagnosis of sporadic schwannomas and neurofibromas depend chiefly on the tumor characteristics (eg, location, size, and growth rate). The broad differential diagnosis of all possible presenting symptoms and signs is vast and beyond the scope of this article.
Lesions of the cerebellopontine angle. Vestibular schwannomas account for a substantial percentage of cerebellopontine angle masses (123). The differential diagnosis of cerebellopontine angle lesions is listed in Table 1.
|
Differential diagnoses |
Characteristics |
|
Lesions originating in the cerebellopontine angle | |
|
Vestibular schwannomas |
Vestibular schwannomas are classically spheric lesions centered around the porus acusticus that deform the canal. They enhance in contrast CT and MRI scans and can sometimes have a dural attachment. |
|
Meningioma |
Meningiomas typically grow as an oval or hemispheric mass rather than as a sphere, and usually have a broad dural attachment. Meningiomas are usually centered away from the porus acusticus, and they rarely enlarge or disturb the canal. Meningiomas demonstrate homogenous enhancement in contrast CT and MRI scans. |
|
Endolymphatic sac tumors |
These are tumors associated with von Hippel-Lindau disease. They appear in the auditory canal. |
|
Epidermoid inclusion cyst |
Lesions are respectively isodense and isointense to the CSF on CT and MRI scans. There may be rim contrast enhancement. |
|
Arachnoid cyst |
Lesions are respectively isodense and isointense to the CSF on CT and MRI scans, without contrast enhancement. |
|
Lesions extending into the cerebellopontine angle | |
|
Metastases |
Most common primary tumors are breast carcinoma, lung carcinoma, and melanoma. |
|
Vascular structure (aneurysm, malformation, or aberrant loop) |
These structures show intravascular enhancement on CT-angiography and MR-angiography, as well as flow void on conventional MRI. |
|
Other central nervous system tumors |
These include exophytic brainstem glioma, ependymoma, choroid plexus papilloma, schwannoma of other cranial nerves (V, VII, IX, X, and XI), jugular foramen paraganglioma, and lipoma. |
|
Infectious diseases |
These include tuberculomas and cysticercosis. |
Other cranial nerve schwannomas. The differential diagnosis of schwannomas arising from cranial nerves other than VIII includes meningiomas, epidermoids, chondromas, chordomas, lipomas, and metastatic lesions.
Spinal schwannomas and neurofibromas. In most cases, spinal schwannomas are indistinguishable from spinal neurofibromas. Tumors to consider in the differential diagnosis of spinal nerve sheath tumors include meningiomas, paragangliomas, ependymomas, and intradural extramedullary metastases.
Peripheral nerve schwannomas and neurofibromas. The differential diagnosis of peripheral nerve schwannomas and neurofibromas consists of other neoplasms such as malignant peripheral nerve sheath tumor, lipomas, metastases, desmoid tumors, granular cell tumors, nerve sheath myxomas, lymphangiomas, and myoblastomas.
Syndrome-related and sporadic schwannomas and neurofibromas. Although the anatomical, perfusion, and diffusion properties of these entities are similar, the existence of many lesions across a nerve and signal heterogeneity are distinguishing markers of syndrome-associated neuromas and schwannomas compared to sporadic tumors (16).
|
• In patients with clinical presentation compatible with a schwannoma or neurofibroma, imaging with a gadolinium contrast MRI is the diagnostic modality of choice. |
Audiometry and neurophysiology for vestibular schwannomas. Clinically, benign vestibular conditions such as Ménière disease can present with symptoms and signs similar to those of vestibular schwannomas (hearing loss, tinnitus, nystagmus, and vertigo). However, a tumoral process can be ruled out by a careful history and examination, followed by appropriate paraclinical tests.
Patients reporting unilateral hearing loss or vertigo can undergo audiometry as a first screening test because only 5% of patients with a vestibular schwannoma will have normal results. Typically, affected patients will demonstrate asymmetrical neurosensorial hearing loss, particularly in the higher frequencies. In addition to audiometry, patients may undergo neurophysiological testing with brainstem-evoked response audiometry, which has a 93% to 98% sensitivity and a 90% specificity for vestibular schwannomas (24).
Despite the favorable diagnostic accuracy of these neurophysiological tests, neuroimaging remains the diagnostic modality of choice.
Neuroimaging of schwannomas and neurofibromas. Radiological findings for sporadic schwannomas and neurofibromas are summarized in Table 2. Benign neurofibromas and malignant peripheral nerve sheath tumor can also at times be differentiated with the use of FDG-PET scanning (31).
|
Tumor |
CT |
T1-weighted MRI |
T2-weighted MRI |
|
Vestibular schwannoma |
Isodense or hypodense to the brain, with homogeneous enhancement; erosion and enlargement of the internal auditory canal in 70% to 90% |
Isointense or slightly hypointense relative to brain and are hyperintense compared to CSF, with diffuse enhancement |
Hyperintense relative to brain, may be isointense to CSF |
|
Trigeminal schwannoma |
Mass with similar characteristics to vestibular schwannoma arising near Meckel cave in the middle fossa, posterior fossa, or both | ||
|
Other cranial nerve schwannoma |
Mass with similar characteristics to vestibular schwannoma arising in the respective territories (eg, hypoglossal canal for XII) | ||
|
Spinal schwannoma and neurofibroma |
Round or lobulated, intradural, extramedullary mass that often compresses the spinal cord |
Isointense or slightly hypointense relative to spinal cord and hyperintense relative to CSF, with diffuse enhancement (may be heterogeneous if there is cystic degeneration, necrosis, or hemorrhage) |
Variable signal intensity (depends on the relative amounts of Antoni A and B zones, cystic degeneration, and hemorrhage) |
|
Peripheral nerve schwannoma and neurofibroma |
Well-demarcated masses isodense to muscle, with some contrast enhancement |
Hypointense with diffuse contrast enhancement |
Hyperintense signal with a characteristic central hypointense region (“target sign”) |
|
Plexiform neurofibroma |
Large multilobulated masses composed of several neurofibromas spreading along nerves and their branches. |
Hypointense with diffuse contrast enhancement |
Hyperintense signal with a characteristic central hypointense region (“target sign”) |
There is significant heterogeneity in the literature regarding the potential associations of tumor structural characteristics on neuroimaging (for instance, size and location) and the degree of neurosensorial hearing loss at presentation among patients with sporadic vestibular schwannomas, suggesting the pathophysiological mechanisms of hearing loss in this population may be multifactorial (06).
Recent work has suggested that diffusion-weighted magnetic resonance imaging may be useful in differentiating plexiform neurofibromas from malignant peripheral nerve sheath tumor among patients with neurofibromatosis type 1 (142; 147; 60). A systematic review and metaanalysis investigating the role of MRI in malignant peripheral nerve sheath tumor diagnosis showed that tumor apparent diffusion coefficient values were highly sensitive (88%) and specific (94%) for malignant peripheral nerve sheath tumor (147). This research suggests that diffusion-weighted MRI characteristics may be superior to conventional morphologic features for identifying malignant transformation, but this requires further investigation in both patients with and without neurofibromatosis type 1.
Biomarkers. Multiple circulating biomarkers have been studied through liquid biopsies to determine if they can be employed for screening and personalized treatment in vestibular schwannoma. Circulating serum VEGF has been suggested as a potential biomarker for selection criteria of patients who would benefit from anti-VEGF therapy. A study also identified nine biomarkers (TNR-R2, MIF, CD30, MCP-3, IL-2R, BLC, TWEAK, eotaxin, and S100B) that are significantly elevated in vestibular schwannoma and can serve as diagnostic biomarkers as well as potential therapeutic targets. Of note, S100B has been associated with tumor growth and MCP-3 with hearing change. They can, therefore, be used to predict disease progression and guide timing of intervention to preserve hearing (136). CSF is thought to be a source of potential biomarkers with ABCA3 and KLF11 having been identified to positively correlate with vestibular schwannoma tumor size, whereas BASP1 and PRDX2 levels were negatively correlated with tumor growth (105). Ki-67 index is also thought to be a useful biomarker for possibility of tumor recurrence following surgical resection. Ki-67 can thereby help detect early tumor recurrence and potential need for early adjuvant therapy (135). At this time, none of these biomarkers have been prospectively validated and, in turn, are not a component of the standard of care.
|
• Overall, schwannomas and neurofibromas can be managed conservatively (with observation), with surgery, radiotherapy, or radiosurgery. | |
|
• In most cases where an intervention is warranted, surgery is the preferred modality and can be curative. | |
|
• There is a limited role to systemic therapy in vestibular schwannomas outside the context of neurofibromatosis type 2-related schwannomatosis. |
Vestibular schwannomas. In general, these tumors can be addressed conservatively, with a watch-and-wait approach, or be subjected to intervention with neurosurgery, fractioned radiotherapy, and radiosurgery (36). The goals are to preserve function, particularly hearing, as long as possible and to minimize risks associated with intervention (149). Data on the success and safety of different interventional treatment modalities continue to emerge, but randomized trials comparing different interventions remain sparse and have limited generalizability (133). It is worth mentioning, however, that upfront radiosurgery was shown to be associated with a more pronounced tumor reduction at 4 years compared to conservative management with observation for patients with newly diagnosed small- and medium-sized vestibular schwannoma (19). Supplementary research on long-term clinical outcomes is required, and these results may help guide treatment choices for vestibular schwannoma patients.
Conservative management (observation). Because these tumors tend to progress slowly, a watch-and-wait approach may be appropriate in carefully selected patients. This strategy is most often employed in patients with significant comorbidities or with important contralateral hearing loss, as well as in elderly patients with small (less than 10 mm) asymptomatic schwannomas (50). A study demonstrated that even large Koos grade 4 vestibular schwannomas that compress the brainstem may be initially appropriate for observation and a wait-and-scan approach, if no significant clinical symptoms of mass effect are present that warrant urgent treatment (114). If hearing loss is an important consideration for patients, early intervention is generally associated with better outcomes than intervention following a period of hearing loss during observation (52).
Candidates who undergo observation should be reimaged at a 6 to 12-months for tumor growth surveillance (09). If there are signs or symptoms of tumor progression, or rapid tumor growth (greater than 2.5 mm/year), intervention should be seriously considered regardless of age (111; 104). There is no specific consensus on the ideal duration of serial neuroimaging in patients who do not demonstrate tumor progression, but several authors commend yearly screening for 5 years, followed by less frequent follow-ups if the tumor remains stable (25; 77).
Surgery. Three surgical approaches are used for vestibular schwannoma resection: (1) suboccipital (retrosigmoid), (2) translabyrinthine (anterosigmoid), and (3) middle fossa (subtemporal). Each has specific advantages to offer, based on selection criteria that include tumor size, depth of internal auditory canal penetration by tumor, hearing status, exposure of the facial nerve, and patient age (129).
|
• The suboccipital approach can be used for any tumor size, regardless of whether hearing preservation is an issue or not. Postoperative headache is most frequent with this strategy. | |
|
• The translabyrinthine approach is mostly employed for large (greater than 3 cm) tumors or for smaller tumors that are located in the deep internal auditory canal. This intervention abolishes ipsilateral hearing and is associated with postoperative CSF leakage. | |
|
• The middle fossa approach is most appropriate for small (less than 1.5 cm) tumors, particularly those outside the internal auditory canal, when hearing preservation is desired. There is an increased risk of facial nerve trauma with this technique. |
Patients undergoing surgery should be monitored for recurrence with yearly neuroimaging. At minimum, they should undergo a control MRI within the year after surgery (25; 78). If there was residual tumor, the minimal duration of follow-up is not clear, with some authors suggesting 3 to 5 years of annual imaging followed by less frequent follow-up in the absence of tumor progression (25; 05).
Patients treated by surgery may also be eligible for simultaneous cochlear implantation, which may restore some degree of ipsilateral hearing following resection (23; 145). Cochlear implantation is preferred over auditory brainstem implantation in vestibular schwannoma patients and offers better hearing outcomes. It offers patients a functioning cochlear nerve, the ability of open set speech discrimination, and an improved quality of life (86).
Radiation therapy. Suitable radiation treatment modalities include stereotactic radiosurgery, stereotactic radiotherapy, proton beam therapy, and fractionated radiation therapy. In the absence of controlled randomized trials comparing these strategies, observational data currently suggest that outcomes are equivalent for all approaches and that the chosen modality should, therefore, depend on patient preferences and center-specific factors, such as expertise and availability (94).
|
• Stereotactic radiosurgery is an interesting option for patients with smaller (less than 3 cm) tumors or for those who are not candidates for surgery. Stereotactic radiosurgery seems to have at least equivalent rates of tumor control and hearing preservation compared to microsurgery, with better facial nerve preservation (03). | |
|
• Stereotactic radiotherapy is an alternative to stereotactic radiosurgery, with radiation delivered over a series of treatment sessions. This is also the modality of adjuvant therapy in patients with subtotal surgical resections. | |
|
• Proton beam therapy is a suitable alternative to the two above-mentioned modalities, although currently there is scant literature available on its outcomes and complications. |
Patients undergoing radiation therapy should be monitored for recurrence with yearly neuroimaging. The minimal duration of follow-up is not clear, with some authors suggesting 10 years of annual imaging followed by repeat imaging every 2 years.
A study attempted to investigate the clinical outcomes of salvage microsurgery following failed stereotactic radiosurgery or fractionated stereotactic radiotherapy in patients with sporadic vestibular schwannoma (81). More than half of patients developed facial paralysis long-term or could not achieve gross total resection. Growth-free survival rates and perioperative complications like postoperative cerebrospinal fluid leak, hydrocephalus, symptomatic stroke, and meningitis were also worse compared to primary microsurgery rates.
The future progress of stereotactic radiosurgery and its implementation into clinical practice will be further driven by continued advances in imaging, targeted therapeutics, artificial intelligence, and biomedical engineering. The combination of pharmacologic and radiation therapies that act synergistically will help reduce the therapeutic dose needed for radiotherapy, thereby minimizing its adverse effects (89).
Other cranial nerve schwannomas. Owing to their rarity, there are no good quality data on the natural history of these tumors. The viable approaches to management for these schwannomas are overall similar to those of vestibular schwannomas, with the exception of proton beam therapy, which has not been extensively studied in this context. Stereotactic radiotherapy has been successfully applied to patients with trigeminal, jugular foramen, and hypoglossal nerves (100), and stereotactic radiosurgery is also proven successful in trigeminal, facial, oculomotor, and jugular foramen schwannomas (84; 57; 119).
Spinal and peripheral schwannomas and neurofibromas. The decision to intervene on spinal and peripheral nerve schwannomas and noncutaneous neurofibromas depends on individual patient factors, such as the degree of symptomatic and functional burden. Cutaneous neurofibromas are not typically removed in the absence of pain, functional impairment, or significant impact on quality of life.
When patients opt to intervene, surgical management is the standard of care. Overall, the outcomes are better for schwannomas than neurofibromas, mostly because the latter operation often implies sacrificing parent and adjacent nerves fascicles.
Systemic therapy. In general, systemic therapy is not frequently utilized for patients with sporadic schwannomas or neurofibromas. Case reports suggest there may be a benefit in using bevacizumab for schwannomas, mandating further research for this agent that has been more extensively studied in the context of NF2-related schwannomatosis (98; 53; 73). VEGF inhibitors mainly seem to have a role in recurrent vestibular schwannoma and vestibular schwannoma already treated with radiosurgery. These cases of vestibular schwannoma appear to have elevated VEGF expression, possibly protecting the endothelial and/or tumor cells during radiation, causing failure of radiosurgery. It was, therefore, suggested that a combination of radiotherapy and anti-VEGF treatment might improve therapeutic outcomes (63). In addition to monoclonal antibodies, immune check point inhibitors also seem to hold promise for vestibular schwannoma like in many other cancers due to reactivation of antitumor cytotoxic lymphocytes (89).
In a phase 2 trial, selumetinib was successful in inducing durable tumor shrinkage and clinical benefit in children with inoperable plexiform neurofibromas caused by neurofibromatosis type 1 (40). Another phase 2 trial showed that cabozantinib was effective in decreasing neurofibromatosis type 1 related inoperable plexiform neurofibroma size and improving patient outcomes, most importantly pain caused by the tumor (33). Finally, a phase II clinical trial demonstrated that the MAPK/ERK kinase inhibitor, mirdametinib, produced a 42% partial response rate and early signs of pain reduction in patients with NF1 and inoperable plexiform neuromas (141). Systemic therapies for neurofibromas and schwannomas are thus likely to be subjected to further research in the coming years, although most likely among populations of individuals with neurofibromatosis. Because the pathophysiological mechanisms driving tumors are the same for patients with sporadic schwannomas and neurofibromas, these therapies could prove useful in this population as well, especially for situations where surgical and radiation therapies are not optimal choices.
The overall prognosis for survival and intact neurologic function for patients with sporadic schwannomas and neurofibromas is relatively good. In most cases, these are benign, slow-growing, encapsulated tumors with a limited capacity for infiltration and destruction of surrounding tissues.
Vestibular schwannomas. Longitudinal studies suggest that interventional management improves patient outcomes (particularly with regards to local tumor control rates and risk of recurrence), but studies that have focused on patient quality of life have not disclosed an advantage of any particular strategy over observation (88).
Conservative management (observation). A majority (59%) of untreated tumors grow at a rate less than 1 mm/year, but some (12%) progress relatively quickly at rates greater than 3 mm/year (02). Tumor growth rate appears to be a significant predictor of subsequent hearing loss in patients with small vestibular schwannomas (less than 25 mm in diameter), with rates greater than 2.5 mm/year associated with poorer hearing prognosis (127). In fact, median time to hearing loss is much quicker (1 to 2 years) in patients with rapid tumor growth than those with patients with slow or absent growth (15 years) (126).
In a metaanalysis comprising 1345 patients followed for a mean follow-up length of 3.2 years, 43% of tumors grew, whereas 57% remained stable or regressed (124). Importantly, 20% of cases ultimately required intervention for significant tumor growth or symptom progression.
Surgery: outcomes. Complete resection is feasible in 97% to 99% of cases (38; 85) and is considered curative (85), with a recurrence rate after gross total resection of only 1% to 2% in most large series. Among patients with incomplete resection, up to 44% will experience tumor regrowth or recurrence, with 26% requiring additional intervention (28). Hearing loss is unlikely to improve after surgery, but most patients retain intact hearing function after surgery. Tumor size appears to be an important determinant of postoperative hearing preservation (101). At 5-year follow-up, 81% of patients with serviceable post-operative hearing maintain functional audition (22). A helpful marker to determine postoperative recurrence and retreatment is the preoperative neutrophil-to-lymphocyte ratio. A preoperative ratio greater than or equal to 2.21 seems to be significantly associated with retreatment after schwannoma resection and can help in the future guidance on treatment decisions (132). Finally, patients with preoperative hydrocephalus do not require permanent shunting after surgery in 78% of cases (97).
Surgery: complications. The most common surgical complications are headache (46%), which can persist after surgery, and CSF leakage (8.5%). Facial nerve trauma is rare (3% to 6%) (38; 85), but an important proportion of patients experience postoperative facial weakness: 70% of patients with large tumors, 50% of those with medium tumors, and 30% of those with small tumors have neuropraxia of the facial nerve after resection (92; 131). Hydrocephalus can occur postoperatively, particularly in the context of large (greater than 3 cm) tumors (97). Mortality is low (0.2%) (128). As is the case with most surgeries, older patients are at higher risk of medical complications following vestibular schwannoma surgery than their younger counterparts (130). Younger age at surgery is independently associated with complete facial nerve recovery, which can assist in intraoperative decision-making regarding extent of resection and postoperative counseling (76).
A study evaluated the quality of life in patients after vestibular schwannoma surgery, which was mostly impaired by postoperative headaches and facial nerve dysfunction. Patients with large tumors seemed to have a noticeably higher risk of postoperative facial nerve paresis, liquorrhea, and lower chances of hearing preservation. Patients with smaller tumors and history of headaches prior to surgery appeared to have more severe and frequent headaches after surgery, which was associated with higher anxiety and tinnitus. Higher anxiety was noted in patients who became deaf after surgery (66).
In terms of hearing preservation, a study showed that patients that demonstrated a hearing preservation advantage are the ones who underwent microsurgical resection within 3 months from time of diagnosis (91).
Radiation therapy: outcomes. Patients undergoing stereotactic radiosurgery with marginal doses of 12 to 13 Gy have good 10 and 15-year local control rates (91 and 89%, respectively), but a quarter of patients (25%) experience long-term hearing loss (08; 45; 44). Risk factors for hearing loss include older age, tumor size and poorer baseline hearing function (107; 93).
Given that stereotactic radiosurgery is associated with iatrogenic hearing loss, a study aimed to examine the level of hearing preservation in 37 patients with serviceable hearing who underwent stereotactic radiosurgery for sporadic intracanalicular schwannoma (51). Hearing was preserved in patients with smaller tumors and in all patients with a tumor volume of 0.05 cm3 or less, lower marginal doses, and with better preoperative hearing. On the other hand, a marginal dose of over 12 Gy and tumor volumes greater than 0.1 cm3 significantly increase the risk of nonserviceable hearing deterioration.
Stereotactic radiotherapy has similar local tumor control success than stereotactic radiosurgery, with 5 and 10-year control rates of 96%, but patients typically receive larger doses of radiation (25 to 60 Gy) (13). Proton beam therapy has a 5-year control rate of 94%, with 33% of patients retaining functional hearing (140).
Radiation therapy: complications. In patients subjected to stereotactic radiosurgery, about 25% experience long-term hearing loss, as mentioned above. In addition, postradiation tumor expansion is observed in 14% of cases (median delay of 9 months). Among these patients, 57% demonstrate subsequent spontaneous tumor regression, 29% remain stable, and 14% have progressive growth that require intervention (99). Facial and trigeminal neuropathy occurs in less than 5% of cases (94), and 2% of patients develop delayed cystic formation (occurring at a median of 6 years after intervention) (45).
Patients undergoing proton beam therapy sometimes require salvation therapy (6%). Facial and trigeminal nerve injury occurs in 9% and 11% of cases, respectively (140).
Other cranial nerve schwannomas. Trigeminal schwannomas, as well as schwannomas of other cranial nerves, have a less favorable prognosis. In general, complete resection is challenging due to inadequate surgical exposure, involvement of the cavernous sinus, encasement of blood vessels, and adherence to the brainstem (125; 07). For trigeminal tumors, the complete resection rate is 70% to 80% in modern series. Recurrence is rare after total extirpation. However, in cases with residual tumor, progression usually occurs within 3 years.
Spinal and peripheral nerve schwannomas and neurofibromas. Complete surgical resection of spinal schwannomas and neurofibromas occurs in 85% to 90% of cases (116; 109). In patients with subtotal removal, recurrence develops in 50% to 55% of cases, but only 18% require a second intervention (117). Patients undergoing peripheral nerve schwannoma resection can experience objective motor improvement (18%) or resolution of subjective symptoms (52%) (56). Complete resection is curative (150). In the context of operated peripheral neurofibromas, resection of tumors located in the brachial plexus is associated with additional postoperative morbidity in 8% to 31% of cases (56).
There is some evidence that a subset of patients with vestibular schwannomas experience rapid tumor growth during pregnancy, but the association has not been robustly documented (118).
In patients with intracranial schwannomas and elevated intracranial pressure, usual precautions are indicated to avoid exacerbations or complications of intracranial hypertension.
Children. To date, a limited number of pediatric patients with sporadic vestibular schwannomas have been reported in the literature (79). Overall, these patients present similarly to adults, but more commonly have symptoms due to mass effect on the posterior fossa structures. For peripheral nerve tumors, patients rarely undergo surgery outside of the context of neurofibromatosis type 1 (151). However, given the known risk of hearing loss after stereotactic radiosurgery, and the high likelihood of post-stereotactic radiosurgery reintervention, microsurgery is the preferred treatment in pediatric patients with vestibular schwannoma (62).
Sex. Multiple sex-specific differences have been reported in the quality of life of patients treated with surgery for vestibular schwannoma. Although energy and balance change similarly in both sexes postoperatively, female patients appear to be significantly more affected by headaches, dizziness, reduced energy, and anxiety. Postoperative women also tended to be more affected by facial palsy and headaches. Despite these differences, general health improved equally or more in female patients compared to males. These should be taken into consideration when counseling patients regarding potential treatment options for vestibular schwannoma (75).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.
Stefania Maraka MD
Dr. Maraka of University of Illinois at Chicago received consulting fees from Alexion Therapeutics and Springworks Therapeutics
See ProfileMariam/Myriam Markouli MD
Dr. Markouli of Boston University has no relevant financial relationships to disclose.
See Profile
Rimas V Lukas MD
Dr. Lukas of Northwestern University Feinberg School of Medicine received honorariums from Jazz Therapeutics, Novocure, and Servier for speaking engagements, honorariums from Cardinal Health, Catalyx, Merck, and Novocure for advisory board membership, research support from BMS as principal investigator, and an honorarium from GT Medical Technologies for DSMB membership.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neuro-Oncology
May. 27, 2026
Neuro-Oncology
May. 27, 2026
Neuro-Oncology
May. 23, 2026
General Child Neurology
May. 12, 2026
Neuropharmacology & Neurotherapeutics
May. 11, 2026
Stroke & Vascular Disorders
May. 03, 2026
Neuro-Oncology
Apr. 30, 2026
Neuropharmacology & Neurotherapeutics
Apr. 23, 2026