



Methylmalonic acidemia
Jun. 01, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Batten disease, or neuronal ceroid lipofuscinosis, constitutes one of the most common groups of inherited childhood-onset neurodegenerative disorders. They currently comprise 13 genetically distinct disorders that differ by causative gene, gene product, and biological process. Neuronal ceroid lipofuscinoses share disease features, including vision loss, epilepsy, cognitive decline, and movement disorders with onset in childhood, adolescence, and even adulthood. Abnormal autofluorescent, electron-dense granules accumulate in the cytoplasm of nerve cells and are associated with selective destruction and loss of neurons in the brain and retina. There are currently 13 different genes and over 360 mutations that underlie these devastating brain disorders. CNS-targeted enzyme replacement therapy is approved for one form of disease: neuronal ceroid lipofuscinosis type 2 (CLN2) disease.
|
• Neuronal ceroid lipofuscinoses, or Batten disease, are common causes of neurodegeneration in childhood. | |
|
• Typical clinical findings include retinopathy leading to blindness, sleep problems, movement disorders, epilepsy, dementia, and premature death. | |
|
• Definitive diagnosis relies on enzymatic assays or DNA testing. | |
|
• Intraventricular cerliponase alfa for ceroid lipofuscinosis type 2 disease is the first and only FDA- and EMA-approved therapy for a specific form of neuronal ceroid lipofuscinosis (CLN2 disease). | |
|
• Kufs disease or adult neuronal ceroid lipofuscinosis can be caused by three different genes and is often misdiagnosed pathologically. |
A detailed history of the classification and the pathological and clinical characteristics has been reviewed (29). Neuronal ceroid lipofuscinosis consists of a group of genetically determined neurodegenerative disorders that affect children and adults of both sexes. The original description of the disorder is credited to Stengel, a Danish physician, who identified four children in a family from a rural village in Norway who had onset of visual failure in their sixth year, followed by progressive intellectual decline and loss of speech. Seizures began at 10 years, and they died in their twenties after remaining in a vegetative state for several years (65).
The visual loss and dementia noted in this disease led to its classification as a form of amaurotic familial idiocy, but an appreciation of the pathological differences, biochemical abnormalities, and genetic defects have established neuronal ceroid lipofuscinosis as a nosologic entity. The eponym Batten disease, now often associated with the juvenile form of the disease or CLN3 disease, is named for Frederick Batten, who, in 1903, described the cerebral and macular changes in two brothers (07). Subsequently, Purkinje cells, gliosis, and the loss of cortical neurons in association with the pathognomonic accumulation of the autofluorescent lipopigments in the remaining neurons were documented and helped to distinguish Batten disease from other syndromes associated with intellectual disability (73; 10; 08). Although the clinical features of the juvenile onset form were delineated in great detail (64; 61), the adult variant was not recognized until 1925 (40). The high prevalence of an infantile onset form of Batten disease in Finland was later identified (57), thereby uncovering the clinical spectrum of neuronal ceroid lipofuscinosis. Other subtypes, such as a variant of late infantile neuronal ceroid lipofuscinosis and a congenital form have been described (41; 22). Advances in molecular genetics have led to the discovery of the gene defects for several of the variants (47). The genetics of this group of disorders demonstrates that they are heterogeneous disorders with common pathologic and clinical features (See Table 1). Neuronal ceroid lipofuscinoses were initially classified by the age at disease onset (eg, infantile, late infantile, juvenile, adult). However, genetic advances have demonstrated that many cases may not follow this strict classification. For example, CLN1 disease patients with juvenile disease onset have been described. Therefore, disease classification based on affected gene is now favored.
|
Gene Name |
Gene Product |
Type of Product |
Typical Age at Onset |
|
CLN1 |
Palmitoyl protein thioesterase (PPT1) |
Enzyme |
Infantile |
|
CLN2 |
Tripeptidyl peptidase (TPP1) |
Enzyme |
Late infantile |
|
CLN3 |
CLN3 |
Transmembrane protein, lysosomal |
Juvenile |
|
CLN4 |
DNAJ heat shock protein family (Hsp40) member C5 (DNAJC5) |
Cysteine string protein |
Adult |
|
CLN5 |
CLN5 |
Soluble lysosomal protein |
Late infantile |
|
CLN6 |
CLN6 |
Transmembrane, ER |
Late infantile |
|
CLN7 |
Major facilitator superfamily domain containing 8 (MFSD8) |
Transmembrane, lysosomal |
Late infantile |
|
CLN8 |
CLN8 |
Transmembrane, ER |
Late infantile |
|
CLN10 |
Cathepsin D (CTSD) |
Enzyme |
Congenital |
|
CLN11 |
Granulin precursor (GRN) |
Soluble cofactor protein |
Adult |
|
CLN12 |
ATPase cation transporting 13A2 (ATP13A2) |
Transmembrane protein |
Juvenile |
|
CLN13 |
Cathepsin F (CTSF) |
Enzyme |
Adult |
|
CLN14 |
Potassium channel tetramerization domain containing 7 (KCTD7) |
Unknown |
Late infantile |
The incidence of neuronal ceroid lipofuscinosis ranges in different countries from 1.3 to 7 per 100,000 live births (49). In one series, neuronal ceroid lipofuscinosis constituted one-quarter of all laboratory diagnoses for neurogenetic diseases (11). The clinical recognition of neuronal ceroid lipofuscinosis still remains difficult, and misdiagnosis is common. However, the inclusion of these disorders on gene panels has improved recognition.
Although the neuronal ceroid lipofuscinoses are distinct diseases, they share similar core clinical features, including vision loss, epilepsy, dementia, and movement disorders. The clinical manifestations and features differentiating the classic forms of the subtypes are shown in Table 2 and in the diagnostic workup section (47). Early development is normal. Early symptoms may include gait abnormalities or other movement disorders such as myoclonus. Ataxia develops in children who are able to walk. Visual loss is a prominent feature in all but some of the adult forms of neuronal ceroid lipofuscinosis or Kufs disease (05; 62). Seizures are common and can vary in age of onset, semiology, and response to antiseizure medications (60). For example, myoclonic seizures are common in CLN2 disease but are infrequent in CLN3 disease (06). The natural histories of CLN2 disease and CLN3 disease have been extensively studied in recent years (02; 50). Neuroimaging parameters are increasingly useful in assessing disease severity and following patients over time (21).
|
CLN1 (Infantile) |
CLN2* (Late infantile) |
CLN3 (Juvenile) |
CLN4 (Adult) |
CLN5 (Variant late infantile) |
CLN6 (Variant late infantile | |
|
Age of onset (years) |
0.5-1.5 |
2-4 |
4-7 |
15-30 |
4-7 |
3-8 |
|
Early development |
Normal |
Normal |
Normal |
Normal |
Normal |
Normal |
|
Visual failure onset (years) |
1-2 |
10 |
4-7 |
Absent |
8-13 |
Variable |
|
Seizure onset (years) |
1-2 |
2-4 |
8-12 |
15-30 |
4-13 |
7-12 |
|
Ataxia |
+ |
+ |
- |
+ |
+ |
+ |
|
Predominant movement disorders |
Myoclonus, hand stereotypies |
Myoclonus |
Parkinsonism |
Myoclonus |
Myoclonus, motor stereotypies |
Myoclonus |
|
Loss of ambulation (years) |
Variable |
5-6 |
14-15 |
Late |
7-15 |
7-11 |
|
Death (years) |
3-12 |
10-15 |
20-30 |
30-45 |
13-30 |
5-12 |
|
Other key findings |
Acquired microcephaly |
Early language regression |
No vision loss |
Adult-onset form also described | ||
|
*Natural history without treatment | ||||||
All forms of the disease lead to seizures, intellectual disability, and early demise. Visual loss is common to all but the adult-onset form of the disease.
A 5-year-old boy with speech delay presented to pediatric neurology with seizures and loss of motor skills. He had an unremarkable birth history. He did not start walking until 18 months of age. By 2 years of age, he had only five words, so he was referred to early intervention. His mom also noted bilateral intermittent hand tremors at 2 years of age.
At 3 years of age, he had his first seizure, which was generalized tonic-clonic in semiology. He was started on levetiracetam. Within 2 months, he started having drop attacks, which were confirmed to be seizures on EEG. Valproic acid was started with good response, although he continued to have drop seizures every few months.
At 4 years of age, he had changes in his gait. He was more unsteady and unable to walk without assistance. He had frequent jerking in his limbs that would lead him to fall. There were no vision concerns at the time.
He had an EEG at 3 years of age that was notable for intermittent discharges over bilateral occipital regions. An MRI brain at 3 years of age was normal. A genetic epilepsy panel was obtained and notable for a pathogenic mutation and a variant of uncertain significance in the TPP1 gene. TPP1 enzyme activity was 3 nmol/hour/mg (normal reference range 86-326 nmol/hour/mg). He was diagnosed with CLN2 disease and initiated on cerliponase alfa therapy. His seizures improved. He did not regain the ability to walk but remained able to crawl.
By 6 years of age, his vision seemed to have worsened because would sit closer to the TV when watching it. On formal ophthalmologic examination, he was noted to have mild optic atrophy with a “bull’s eye” maculopathy with retinal pigment epithelial (RPE) stippling in the perifovea and RPE mottling. On optical coherence tomography, he had small areas of residual photoreceptors and loss of normal definitions of the inner retina. These ophthalmologic findings are consistent with CLN2-associated retinal dystrophy.
All forms of Batten disease are inherited. Most forms are transmitted as autosomal recessive traits, except for CLN4 disease, which in some cases, may also show a dominant pattern. Although autofluorescent lipopigment accumulation is characteristic to all the forms, it occurs due to a variety of genetic defects. The known gene loci are listed in Table 1. The pathogenesis and pathophysiology for the cellular dysfunction and pathologic changes is unclear; however, our understanding is improving as genetic characterization progresses.
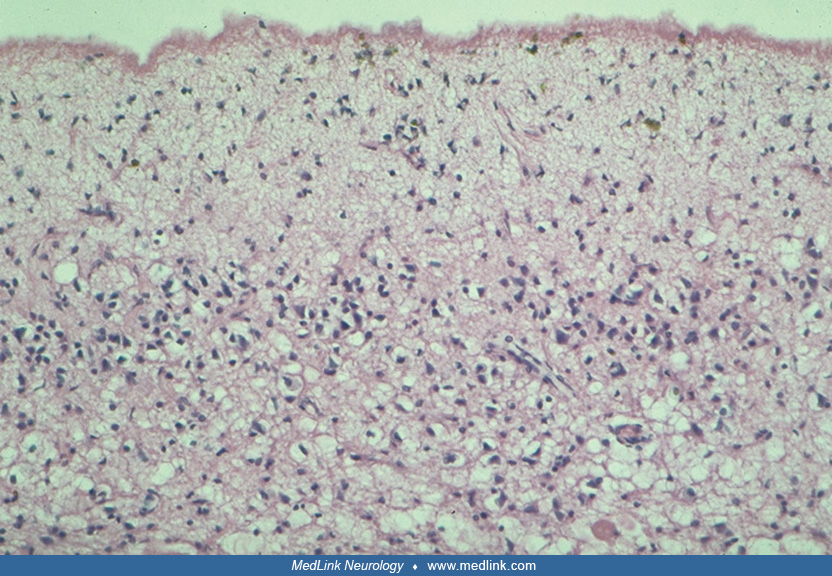
Thirteen genetically distinct neuronal ceroid lipofuscinosis variants, categorized by age of onset and pathological features, have been identified. Ceroid lipofuscinosis-causing mutated proteins represent soluble lysosomal enzymes, polytopic membrane proteins localized in lysosomes or in the endoplasmic reticulum, or synaptic vesicle associated proteins (35). The relationship between genetic defects associated with the major forms of neuronal ceroid lipofuscinosis, the accumulation of storage material, and tissue dysfunction or damage is still unknown (49). Furthermore, all individuals with neuronal ceroid lipofuscinosis manifest lysosomal storage in many tissues and organs, but severe degeneration and cell loss involve mostly neuronal cells. Thus, it appears that neuronal ceroid lipofuscinosis proteins may be most critical for the metabolism of neurons (49). The early and widespread accumulation of autofluorescent lipopigment inclusions in various organs is a characteristic feature of human and animal models of neuronal ceroid lipofuscinosis. The inclusions, which under the electron microscope appear as fingerprint profiles, as curvilinear, or as granulomatous bodies, may not always be detected or correctly interpreted, leading to missed diagnosis (77; 15).
These inclusions may also be seen in the mucopolysaccharidoses and other disorders (25; 79; 81; 54). The largest accumulation occurs in neurons and retinal cells. The lipopigment accumulates at a much earlier age than lipofuscin of aging and differs from the lipopigment in that it contains the ceroid predominantly. Pronounced and selective neuronal vulnerability and total loss of small pigment-laden stellate cells have been demonstrated in the cortex of patients with juvenile neuronal ceroid lipofuscinosis and adult neuronal ceroid lipofuscinosis (13). Spindle-shaped swellings in the axon hillock and, particularly, in the initial segment of pyramidal cells precede other signs of neuronal degeneration. Decreased numbers of spines along apical dendrites are also noted. Astrocytes show storage material in the mossy fiber system (12). The timing and extent of neuronal death vary greatly among the different forms of neuronal ceroid lipofuscinosis and appear to be unrelated to the degree of intraneuronal storage.
This early and severe neuronal loss in infantile neuronal ceroid lipofuscinosis has led to the consideration of neurotoxic mechanisms (28) and to the possible importance of the enzyme in the postnatal maturation and survival of cortical neurons (30). Interestingly, Walkley noted that the GABAergic cells bear the major burden in this disease as evidenced by a prominent loss of GABAergic synapses and neurons (74). This is attributed to the high metabolic rates of GABAergic cells, which become more vulnerable from intraneuronal storage. The susceptibility of GABAergic neurons seems to be confirmed by more recent data from human cellular models (26).
Pathological studies of the retina in the late infantile and juvenile forms demonstrate curvilinear profiles and multimembranous bodies. The conjunctivae show similar inclusions. In both forms of the disease, the retinal destruction starts at the photoreceptor and outer retinal levels and progresses from the macular area to the periphery (67).
In brains from each of the childhood forms of neuronal ceroid lipofuscinosis and the ovine model, a 10-fold to 20-fold increase in phosphorylated dolichols has been identified. This is higher than the 2-fold to 5-fold increase noted in different lipidoses and elderly human subjects (27; 53). The mechanism by which phosphorylated dolichols accumulate in neuronal ceroid lipofuscinosis is presently unknown, but these compounds account for the photoacoustic spectroscopy-positive component on histopathology.
Many biochemical abnormalities have been reported, but with molecular diagnoses of some of the disorders, enzymatic abnormalities are unlikely to be the primary defect. The gene product in CLN3 disease localizes to the Golgi apparatus in human fibroblast, and cell line cultures suggest a role in protein packaging or transport (39).
Since 1995, molecular genetic studies have identified over 360 mutations in 13 different genes underlying the various established human forms of neuronal ceroid lipofuscinoses (38). See the NCL Mutation and Patient Database. The defects in CLN1 disease have been identified in the palmitoyl-protein thioesterase gene in Finnish and non-Finnish patients with this disease (71). The mutation is a transversion from adenine to thymidine at the 364 position. Palmitoyl-protein thioesterase is normally incorporated into lysosomes where it presumably acts to remove fatty acid palmitate residues from proteins. The mutated form of the protein does not become incorporated into lysosomes in vitro (31). Palmitoyl-protein thioesterase deficiency could be demonstrated in lymphoblasts, and most of the mutant polypeptide appears to be trapped in endoplasmic reticulum. The absence of palmitoyl protein thioesterase in lysosomes may lead to the accumulation of undigested product. Palmitoyl protein thioesterase activity was undetectable in brain tissue from patients with infantile neuronal ceroid lipofuscinosis but was normal in patients with juvenile neuronal ceroid lipofuscinosis (71). A variant of early infantile neuronal ceroid lipofuscinosis (CLN6) was found in the Gypsy and Indian population (66).
The genetic defect CLN2 disease has been mapped to chromosome 11p15, and the gene has been sequenced (45). It consists of 13 exons and 12 introns spanning 6.65 kb. Four mutations have been identified but when screened in 16 probands; an intronic mutation, T523 G-->C was detected in 56% of cases (34% of chromosomes), and a nonsense mutation 636 C-->T was found in 31% of cases (19% of chromosomes). Two other previously described missense mutations, 1107 T-->C and 1108 G-->A, were not identified in any of the 16 patients (82). The gene product has been characterized as a lysosomal enzyme, tripeptidyl peptidase I, that cleaves tripeptides from the N-terminus of polypeptides (72). Fibroblasts from patients with late infantile neuronal ceroid lipofuscinosis have less than 5% of the normal tripeptidyl peptidase I activity and are defective in degrading short polypeptides.
A variant form of late-infantile neuronal ceroid lipofuscinosis has an age of presentation similar to classic CLN2 disease but has a clinical course more consistent with CLN3 disease. Tissue from these patients manifests granular osmiophilic deposits when examined by electron microscopy. These patients have normal levels of subunit c in urine and deficient (less than 10% of normal) activity of palmitoyl-protein thioesterase (78), the enzymatic defect in infantile neuronal ceroid lipofuscinosis (CLN1). Patients with granular osmiophilic deposits had mutations in the palmitoyl-protein thioesterase gene that were different from those in patients with infantile neuronal ceroid lipofuscinosis (80). These were both missense and nonsense mutations. In CLN2 disease, there are thus far 131 unique variants from 389 individuals (717 alleles) collected from the literature review, public databases, and laboratory communications (24). Previously unrecorded individuals were added to the UCL TPP1-specific database. Two known pathogenic variants, c.509-1 G>C and c.622 C>T (p.(Arg208*)), collectively occur in 60% of affected individuals in the sample, and account for 50% of disease-associated alleles (24).
The gene for the Finnish variant of late infantile neuronal ceroid lipofuscinosis (CLN5) has been identified through positional cloning on chromosome 13. The gene encodes a novel transmembrane protein in which deletions, nonsense mutations, and missense mutations have been found in patients with this disorder (58).
The CLN3 disease gene has been localized to chromosome 16p12 (03). The so-called 56 chromosome haplotype is shared by 73% of juvenile neuronal ceroid lipofuscinosis chromosomes. These chromosomes contain a 1-kb genomic deletion in the candidate gene. Two separate deletions and one point mutation in three other unrelated families confirmed the candidate gene as the juvenile neuronal ceroid lipofuscinosis gene that encodes a novel 438 amino acid protein. Molecular modeling and homology searches suggest a strong evolutionary conservation of function based on high homology with a yeast protein and possible localization to the mitochondrial membrane (32) where it could function in the transport of other proteins, specifically subunits of the mitochondrial ATP synthase complex. Subunit c of mitochondrial ATP synthase accumulates in lysosomes of juvenile neuronal ceroid lipofuscinosis and late-infantile neuronal ceroid lipofuscinosis patients. In patients with late-infantile neuronal ceroid lipofuscinosis, the accumulation of subunit c is associated with decreased degradation of subunit c within lysosomes due to structural alterations of subunit c and to decreased proteolysis within the lysosomes (51). Interestingly, CLN3 mutations underlying juvenile neuronal ceroid lipofuscinosis cause significantly reduced levels of Palmitoyl-protein thioesterases-1 (Ppt1)-protein and Ppt1-enzyme activity in the lysosome (04). Two other variants of late-infantile and juvenile neuronal ceroid lipofuscinosis (CLN7 and CLN8) exist as well (23). CLN8 has a congenital form as well (52). Adult onset neuronal ceroid lipofuscinosis or Kufs disease may have both recessive and dominant inherited forms. Recessive adult-onset neuronal ceroid lipofuscinosis (ANCL) has been divided into two overlapping clinical subtypes presenting predominantly as (1) progressive myoclonus epilepsy with dementia, ataxia, and late-onset pyramidal and extrapyramidal signs (type A, CLN6 disease) or (2) progressive behavioral abnormalities and dementia, which may be associated with motor dysfunction, ataxia, extrapyramidal signs, and suprabulbar signs (type B). Some adult-onset neuronal ceroid lipofuscinosis families with autosomal dominant inheritance are referred to as Parry disease (69). Kufs disease type A is caused by CLN6 or DNAJC5. Cathepsin F (CTSF) was linked to Kufs disease type A or B (CLN13) (69). Pathological diagnosis is particularly difficult and is often confused with other forms of dementia (09). CLN10 disease is a very rare and severe congenital form of neuronal ceroid lipofuscinosis caused by mutations in the lysosomal aspartic protease cathepsin D gene (CTSD) (47; 70). This is the earliest onset form of neuronal ceroid lipofuscinosis. It presents in the neonatal period with microcephaly due to brain atrophy, absence of neonatal reflexes, and respiratory insufficiency (47; 70). Hypertrophic cardiomyopathy may be present as previously reported in CLN2 and CLN3 diseases (20).
The mechanism of disease and the reason these ubiquitous proteins cause disease only in the brain is unclear. At least three of the dysfunctional proteins (CLN1 to CLN3) reside in lysosomes, but the mechanism by which they cause neuronal death is not known (75). Some investigators suggest that a common mechanism is mitochondrial malfunction (33). It is thought that CLN3 protein is involved in late endosomal/lysosomal membrane transport (68). It is likely that accumulation of proteins is toxic to neurons and, in particular, to synaptic structure and function (18; 37).
The neuronal ceroid lipofuscinosis group of disorders have an incidence of 1 to 3 per 100,000 and a prevalence of 2 to 4 per 1,000,000 (60). CLN3 disease is the most common form in Western countries whereas CLN2 disease is the most common form in Southern Europe and the Mediterranean. Updated epidemiologic information is likely as genetic testing becomes more widespread. An International NCL Database (NCT04613089) was started in 2020 and is collecting data on patients with neuronal ceroid lipofuscinosis from 19 countries.
Although no preventative measures are available for any of the variants of neuronal ceroid lipofuscinosis, elucidation of the genetic basis for many of the neuronal ceroid lipofuscinoses makes carrier detection and prenatal diagnosis more feasible.
The differential diagnosis of neuronal ceroid lipofuscinosis is difficult and misleading due to the various subtypes, increasing number of variants, and the commonality of symptoms with many other neurodegenerative disorders. Visual loss may be missed in a child with seizures and loss of cognitive skills. The electron-microscopic evaluation of the skin could miss the inclusions, and although rectal biopsy has a higher rate of diagnostic success, it is not commonly studied. The adult form of the disease has both autosomal recessive and dominant modes of inheritance. This form may not be readily recognized due to its rarity and to the absence of visual loss, which is a cardinal symptom in the other subtypes.
A potential diagnosis of neuronal ceroid lipofuscinosis should be considered in a child with afebrile seizures between 2 to 4 years of age with delayed expressive language, rapid progression of loss of vision in childhood, or a progressive movement disorder with cerebellar atrophy (60). In CLN3 disease, vision loss often precedes the onset of seizures or motor symptoms. Genetic testing through gene panels (eg, epilepsy panels), whole exome sequencing, or whole genome sequencing should be undertaken. If the diagnosis remains uncertain but a neuronal ceroid lipofuscinosis is considered, enzyme analysis for CTSD, CTSF, PPT1, and TPP1 should be done. Although ultrastructural examination was commonly employed for diagnoses in the past, it is now reserved for patients in whom genetic testing cannot be completed or if the results remain unclear. Ancillary testing, such as MRI brain and EEG, may also be helpful.
Besides traditional MRI, patients with CLN3 disease were found to have globally decreased anisotropy and increased diffusivity (56). In the same study, there was also increased diffusivity and decreased anisotropy in areas such as the corona radiata and posterior thalamic radiation (56).
Photoparoxysmal response on intermittent photic stimulation was present from the first EEG in 93% (13 of 14) of patients with CLN2 disease (63). Background slowing and epileptiform discharges are common in all types of neuronal ceroid lipofuscinosis. For the disorders with abnormal palmitoyl-protein thioesterase-I activity, enzymatic assays have been shown to be reliable using immortalized lymphoblasts and postmortem brains (17).
Nonspecific therapy. Current treatment consists of symptomatic relief of seizures, feeding problems, behavior disorders, movement disorders, and visual loss. In a small study of 16 patients, lamotrigine was effective in decreasing seizure frequency as part of combination therapy (76). Bone marrow transplantation in affected dogs has been ineffective (36). A report of one patient with late-infantile neuronal ceroid lipofuscinosis and of another with Juvenile neuronal ceroid lipofuscinosis who received bone marrow transplant, did not report any improvement (42). Autoantibodies to glutamic acid decarboxylase 65 (GAD65) were detected in the sera of patients with juvenile neuronal ceroid lipofuscinosis and in CLN3 knockout mice (16). These antibodies occur mostly late in the disease, and efforts to lower them have not yielded clear results (01). Cysteamine bitartrate and N-acetylcysteine are potent antioxidants and scavengers of reactive oxygen species (43). A pilot study using these two compounds was associated with decreased patient irritability, delay of isoelectric EEG, and depletion of granular osmiophilic deposits in infantile neuronal lipofuscinosis (43). Gene therapy may be the best way for the future (14; 48).
Specific therapy. In April 2017, the FDA approved the first specific therapy for late infantile neuronal ceroid lipofuscinosis type 2 (CLN2). Cerliponase alfa is a recombinant form of human TPP1, the enzyme deficient in patients with CLN2 disease (44). It is administered into the CSF by infusion via a specific surgically implanted reservoir and catheter in the head (intraventricular access device). Collaboration with a treatment center familiar with cerliponase alfa and development of a multidisciplinary team to help create a treatment process for patients are recommended (19).
The efficacy of Brineura was established in a non-randomized, single-arm dose escalation clinical study in 24 symptomatic pediatric patients with CLN2 disease and compared to 42 untreated patients with CLN2 disease from a natural history cohort (an independent historical control group) who were at least three years old and had motor or language symptoms (59; 50; 44). It is likely that this product is just the first of specific therapies that will be developed in the future for the various forms of neuronal ceroid lipofuscinosis (48). The development of this therapy emphasizes the need of quantitative natural history studies in rare diseases (50). Newborn screening for these diseases will become increasingly important with the development of disease modifying therapies (34).
The pipeline for emerging specific therapies in neuronal ceroid lipofuscinosis is robust. Potential targeted therapies include small molecule drugs, large molecule drugs (eg, enzyme replacement therapy), and gene therapies (46). Clinical trials are ongoing, but none of these potential treatments are FDA approved.
Cerliponase alfa has been shown to slow the decline of motor and language function in children with CLN2 disease. Long-term effects of cerliponase alfa treatment are under investigation but are currently unknown. More common adverse events related to treatment include convulsions, fever, emesis, and hypersensitivity reactions (59). More serious adverse events include failure of the device and device infection.
A summary of published cases of anesthesia in patients with neuronal ceroid lipofuscinosis was published (55). There are no specific risks; however, the neurologic condition should be taken into account and short-acting anesthetic agents should ensure rapid recovery after surgery (55).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.
Jennifer A Vermilion MD
Dr. Vermilion of University of Rochester Medical Center received a consulting fee from Biomarin.
See Profile
AHM M Huq MD PhD
Dr. Huq of Wayne State University has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neurogenetic Disorders
Jun. 01, 2026
Neurogenetic Disorders
May. 08, 2026
Neurogenetic Disorders
Apr. 30, 2026
Neuropharmacology & Neurotherapeutics
Apr. 23, 2026
Neurogenetic Disorders
Apr. 14, 2026
Neurogenetic Disorders
Apr. 14, 2026
Neurogenetic Disorders
Apr. 14, 2026
Neurogenetic Disorders
Apr. 14, 2026