





Sleep Disorders
Sudden infant death syndrome
Jul. 05, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) is an autosomal dominant genetic disease that induces recurrent subcortical ischemic strokes and ultimately leads to severe disability and death. Molecular studies have revealed important genotype-phenotype relationships in CADASIL. In this updated article, the authors discuss advances in the pathogenesis and pathophysiology of this disorder, the differential diagnosis and diagnostic approach, and experimental approaches utilizing immunotherapy to treat this important genetically induced cause of stroke.
|
• Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) is a genetically determined disorder that leads to early transient ischemic attacks and strokes. | |
|
• Initial symptoms of CADASIL may include migraine headaches or psychiatric disturbance. | |
|
• MRI scan of the brain is always abnormal in symptomatic patients and shows signs of small deep infarcts and leukoencephalopathy. | |
|
• CADASIL is an autosomal dominant disorder caused by mutation in Notch3 on chromosome 19, and genetic testing is commercially available for this disorder. | |
|
• Although the disease manifests itself solely as brain dysfunction, the vasculopathy of CADASIL is systemic, thus, providing the opportunity for diagnostic biopsies from skin, muscle, or peripheral nerve. |
In 1977, the first reports were published of a hereditary disorder characterized by recurrent subcortical ischemic strokes and stepwise progression of neurologic deficits leading to dementia, pseudobulbar palsy, and severe disability (98; 99). Since then, the disorder has been referred to by several names, including "hereditary multi-infarct dementia" and "familial sclerosing vasculopathy" (24; 97). In the 1990s, the disorder came to be known formally by the awkward, but clinically accurate designation "cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy." It was immediately recognized that a more user-friendly working nomenclature was needed, and the acronym "CADASIL" emerged (103; 96). The acronym is now more commonly used (and probably more commonly recognized) than the full name of the disorder. Therefore, as is true of virtually all manuscripts, book chapters, and discussions of this subject, this review will use the acronym throughout its text.
|
• A principal characteristic of CADASIL is recurrent ischemic strokes, usually beginning in mid-adulthood. | |
|
• The strokes often occur in the absence of other stroke risk factors. | |
|
• A history of migraine headaches with aura and psychiatric disturbance often precedes the onset of strokes by multiple years. | |
|
• CADASIL is marked by cognitive decline that progresses to dementia. | |
|
• Diffuse white matter lesions and subcortical infarcts are evident on neuroimaging. |
CADASIL is characterized clinically by recurrent subcortical ischemic strokes, usually beginning in mid-adulthood, with a progression to pseudobulbar palsy and dementia (102). The recurrent strokes usually begin in the fifth or early sixth decades and typically occur in the conspicuous absence of traditional cerebrovascular risk factors (18).
The principal neurologic disturbance in most patients is cognitive decline. This usually begins with executive dysfunction, which is often present in the fourth or fifth decades of life. Visuospatial and reasoning abilities deteriorate with age, usually after the age of 50 years. During the seventh decade of life, skills in all cognitive domains deteriorate and are typically diffusely impaired (15).
Although symptoms of CADASIL often begin to appear by early adulthood, there is considerable phenotypic variability in timing of onset and severity of disease (32). Some patients who carry the CADASIL gene mutation can have no clinical, neuroimaging, or laboratory evidence of disease, even into the sixth decade of life (47). This variability of onset and severity appears to be due largely to location of the gene mutation, which will be explained in more detail below.
Although subcortical stroke or transient ischemic attack is the initial symptom in most affected individuals, the first symptom may be migraine-like headache or psychiatric disturbance, including depressive or manic episodes (12). Rarely, the initial symptom may be ataxia (88). MRI of the brain is always abnormal in symptomatic patients and consistently shows signs of small deep infarcts and a striking leukoencephalopathy (63). Such MRI abnormalities may be detectable in affected individuals prior to the onset of any symptoms (12). In addition to white matter abnormalities, cortical lesions can also be detected by MRI scan in a substantial portion of patients with CADASIL. However, virtually all cases of these cortical lesions are associated with deep progressive signal changes within the underlying white matter, thus suggesting that the cortical changes actually reflect a progressive extension of white matter disease (74).
Although the great majority of patients with CADASIL present with symptoms referable to the brain, some present with spinal cord involvement. Transient ischemic attack and strokes involving penetrating arteries of the spinal cord, producing isolated weakness of the arms or legs, have been the presenting symptoms. In all of these patients, however, neuroimaging studies of the brain have revealed extensive cerebral white matter involvement, indicating that cerebral pathology is present in these patients even if it has not yet manifested clinically (55).
Although the disease usually has its onset during adulthood, multiple cases have been reported of CADASIL in the pediatric age group. To date, the youngest patient with genetically confirmed CADASIL has been 3 years old (08). This toddler presented with global developmental delay and hyperactivity. Among pediatric-onset cases, adolescence is the most common age of presentation. Young patients with CADASIL may have recurrent subcortical strokes as their presenting sign, but migraine headaches and deficits in attention and memory seem to be more common (40). When migraine headache is a presenting symptom of CADASIL, features that are atypical of migraine are almost always also present. These atypical migraine features include unusual headache patterns, such as aura without headache, prolonged aura, or atypical white matter lesions on MRI (22).
The clinical manifestations of CADASIL may be somewhat different in women than in men (44). In particular, before 51 years of age, migraine with aura is more prevalent in women with CADASIL than in men with the disease. Conversely, in this same age group, stroke events and cerebral atrophy are more prevalent among men. Both before and after 51 years of age, apathy is twice as prevalent in male as in female patients. These differences in disease characteristics exist despite the fact that the prevalence of transient ischemic attack, age at first stroke, and number of stroke events does not differ according to gender. The differences in clinical manifestations of CADASIL between the two sexes are likely due to sex hormones. In particular, estrogen and ovarian hormones, which circulate at high levels in premenopausal women, promote spreading depression and migraine headaches. At the same time, these hormones increase cerebral blood and vasoreactivity, and exert anti-inflammatory, antioxidant, and antiapoptotic effects. Thus, it is likely that sex hormone profiles worsen CADASIL’s migraine effects in women and stroke effects in men (39).
Although cognitive decline, pseudobulbar signs, and other evidence of stroke are the most common signs of CADASIL, some patients have parkinsonian symptoms as a prominent feature (91). The parkinsonism in CADASIL is typically late in onset and includes akinesia, rigidity, and postural instability. Resting tremor and asymmetry are typically absent. In CADASIL, the parkinsonian symptoms do not respond to levodopa and are consistent with a vascular parkinsonism (91).
Because there is substantial phenotypic variability within CADASIL, recent efforts have focused on the development of systems to grade illness severity. One multicenter retrospective study introduced a CADASIL grading system in which patients were categorized into groups according to symptom severity: grade 0 (asymptomatic), grade 1 (migraine only), grade 2 (stroke, TIA, or MCI), grade 3 (gait assistance or dementia), and grade 4 (bedbound or end stage). Higher severity grade was associated with an increased number of vascular risk factors, older age, hypertension, and diabetes (03). This system and others like it will be useful for categorizing patients with CADASIL for observational and interventional studies.
The prognosis for patients with CADASIL is grim. Following the onset of symptoms, the clinical course is marked by a stepwise deterioration in sensory motor function, development of a pseudobulbar palsy, and a relentlessly progressive dementia (13). A study found that the first attack of migraine headache occurred at a mean age of 38.1 years, that transient ischemic attacks and strokes began at 49.3 years, and that the mean age of demented subjects was 58.2 years (20; 21). Most patients with CADASIL die within 10 to 20 years of clinical onset. There have been no studies demonstrating that the poor prognosis of CADASIL can be altered by therapeutic intervention.
Although the disease is always progressive, certain demographic, clinical, and magnetic imaging factors have been identified that can predict specific forms of impending clinical deterioration (17). In particular, cigarette smoking, the presence of lacunes, and evidence of brain atrophy are each independent predictors of impending stroke. The presence of incident lacunes on MRI scan are associated with incident stroke and with cognitive and executive function losses (71). Gait disturbance, dementia, and brain atrophy predict earlier progression toward more severe disability. In addition, cigarette smoking and brain atrophy predict an earlier dementia. Thus, several factors can forewarn looming clinical deterioration, but among them, active smoking is the only modifiable risk factor.
Although the prognosis for CADASIL is poor, the severity and age-of-onset can vary substantially among patients and appear to depend strongly on the pathogenic variant position within the NOTCH3 gene. In particular, pathogenic variants within the EGFr (epidermal growth factor repeat) domains 16 portend a much worse prognosis than pathogenic variants within EGFr 7-34. Indeed, patients with an EGFr 1-6 pathogenic variant have a 12-year earlier onset of stroke, higher white matter hyperintensity volumes, and shorter survival than those with an EGFr 7-34 pathogenic variant (95). A study has shown that people with pathogenic variants within EGFr 1-6 are more than twice as likely to have a stroke than those with a pathogenic variant within EGFr 7-34. The study further showed that this increased vulnerability accompanying the EGFr 1-6 mutation location leads to earlier dementia and dependence, both of which are tied to development of more damaging ischemic tissue lesions (32). Thus, there is a strong genotype-phenotype relationship with regard to disease kinetics and prognosis in CADASIL.
The complications of CADASIL are similar to those of any other condition producing stroke and dementia. However, one complication that is particularly common and serious in CADASIL is intracerebral hemorrhage. The vasculopathy of CADASIL renders cerebral blood vessels particularly prone to rupture. For this reason, anticoagulants should be avoided in CADASIL if possible.
The patient was healthy until 52 years of age, when he presented with a 2-month history of episodic word-finding problems, difficulty understanding written language, reduced initiative, increased lethargy, and right arm and leg numbness. Three months later, he had an acute onset of right hemiparesis and increased difficulty with expressive language. He was admitted to a hospital, where his symptoms progressed over the course of hours to global aphasia, worsened right hemiparesis, and incontinence. An MRI of the brain revealed multiple old subcortical lacunar infarctions and diffuse white matter abnormalities. Daily aspirin was prescribed, but his condition worsened over several days, and warfarin was added. He was discharged on warfarin and aspirin therapy. Over the course of the next several months, he had a series of minor strokes. An MRI of the brain 6 months after presentation revealed marked white matter changes in both hemispheres consistent with multiple small vessel occlusions.
Over the course of the next 18 months, his condition worsened in a stepwise fashion coincident with minor strokes, and he became increasingly demented.
On examination at 54 years of age, he was obviously mentally impaired but appeared otherwise healthy. Vital signs were all normal, including a blood pressure of 101 systolic over 63 diastolic. He was alert, but oriented only to name and not to place or date. He had little spontaneous speech and made frequent paraphasic errors. He had a marked pseudobulbar affect. Cranial nerves were intact with the exception of a mild right lower facial paresis. Funduscopic examination was normal. He had a right hemiparesis with more involvement of the upper extremity than of the lower extremity. Muscle stretch reflexes were hyperactive globally, but more so on the right, with a Babinski sign present on the right. Sensory examination was grossly intact. The general physical examination was unremarkable. No murmurs or bruits were heard over the head or neck.
Laboratory evaluation included a normal complete blood count with differential, prothrombin time, partial thromboplastin time, international normalized ratio, and platelet count. Serum electrolytes, liver enzymes, blood urea nitrogen, creatinine, and ammonia were normal. Blood lipid profile and thyroid function tests were normal. Serum VDRL was nonreactive. Serum protein electrophoresis and amino acid analysis, including homocysteine, were normal. Antinuclear antibodies, Lyme antibody titer, and anticardiolipin antibodies were all negative. Cerebral angiography revealed no large vessel disease and no evidence of vasculitis or other small vessel disease. Echocardiogram showed normal left ventricular function, no intracardiac thrombus, and structurally and functionally normal valves. Duplex carotid ultrasound was unremarkable. EEG was moderately abnormal with excessive bitemporal and diffuse theta-delta slowing indicative of diffuse cerebral dysfunction.
The family history was remarkable, as demonstrated by the pedigree.
Multiple family members, including four of the patient's five siblings, his father, his paternal grandmother, and his paternal great-grandfather, had transient ischemic attacks or strokes at relatively young ages and developed dementia. This condition, in this family, had passed through five known generations and appeared to have an autosomal dominant pattern of inheritance.
The patient continued to decline in a stepwise fashion over the course of the next decade and died at 64 years of age. By the time of his death, he was markedly demented and quadriparetic. Two years after his death, genetic testing for CADASIL became commercially available. A younger sibling who had suffered recurrent transient ischemic attacks and strokes beginning at 53 years of age underwent genetic testing, which revealed a mutation in the Notch3 gene, thus, confirming the diagnosis of CADASIL.
|
• CADASIL is an autosomal dominant genetic disease due to pathogenic variants in the NOTCH3 gene. | |
|
• Notch3 is a transmembrane receptor that is expressed by vascular smooth muscle cells and pericytes in vasculature throughout the body. | |
|
• Notch3 mutations affect 1 of 34 extracellular domain epidermal growth factor repeats of the Notch3 protein by either removing or adding a cysteine residue. | |
|
• This altered Notch3 protein leads to deposits of granular osmiophilic material near blood vessels and degeneration of the vascular smooth muscle layer of blood vessels. | |
|
• The pathogenic cascade eventually leads to strokes through unknown mechanisms. |
CADASIL is an autosomal dominant genetic disease caused by mutations in the Notch3 gene, located on chromosome 19 (48). Notch3 is a large gene of 33 coding exons. Most of the mutations in CADASIL occur in exon 3 or 4, which encode the first five epidermal growth factor-like repeats (62).
Although CADASIL is directly caused by the mutation of a specific gene (Notch3), there is variability in the onset, severity, and progression of the disease from case to case, even among patients carrying the same Notch3 mutation. This suggests that other factors also play a role in the etiology. One strong possibility is that there are genetic modifiers of the disease. To explore this possibility, a genome-wide association study was completed to identify quantitative trait loci that influence CADASIL. In particular, quantitative trait loci were sought for the phenotypic variance in the volume of white matter hyperintensities on MRI scan in patients with CADASIL. Indeed, the researchers found a polygenic score associated with the white matter hyperintensity volume in patients with CADASIL. No single gene or gene region reached genome-wide significance. However, the results suggest that multiple common genetic variants, each with a small effect size, influence the white matter hyperintensity burden (and probably the severity of clinical disease) in CADASIL (86).
Pathologic examination of the brain in CADASIL consistently reveals multiple small deep infarctions throughout the subcortical white matter, typically accompanied by diffuse myelin loss and pallor of the hemispheric white matter (96). The cortex and U-fibers are relatively spared. These pathologic changes in the subcortical white matter not only reflect the recurrent episodes of stroke, but also, presumably, underlie the dementia that accompanies the syndrome.
Because both demyelination and multiple lacunar infarctions can induce dementia and are present in CADASIL, several studies have sought to determine which of these two processes is most responsible for the cognitive deficits in CADASIL (70; 104). Both studies utilized MRI scans to quantify demyelination and lacunar load. The volume of white matter hyperintensity on T2-weighted images (indicative of demyelination) and the number of parenchymal defects with signal intensity corresponding to cerebrospinal fluid (indicative of lacunes) were compared with neuropsychological test scores (indicative of dementia). Both studies found that the volume of lacunar lesions was strongly correlated with degree of cognitive dysfunction whereas the extent of white matter hyperintensities was not. These studies suggest that the ischemic process leading to lacunar infarction is more important than demyelination in the pathogenesis of CADASIL.
Pathologically, the primary disease process is a systemic vasculopathy.
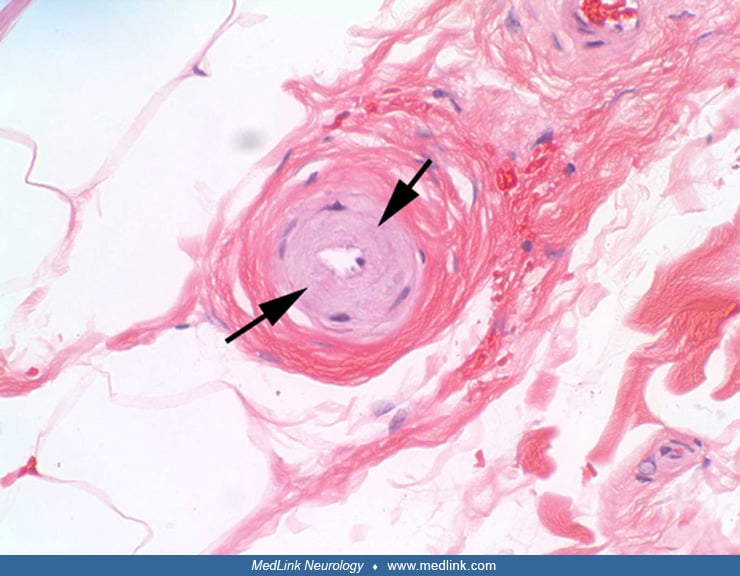
Nonamyloid angiopathy is characterized by the presence of granular, electron-dense, osmiophilic, eosinophilic material within the media of small arteries and arterioles (45). Over time, deposition of this granular material replaces the smooth muscle cells of the arterial media in small vessels, resulting in a loss of responsiveness of the vessel to extrinsic factors (07; 64; 19; 90; 52). The resulting concentric thickening of the arterial wall also induces a narrowing of the vessel lumen, especially in the long penetrating cerebral arteries supplying white matter (78). These two factors presumably lead to chronic arterial insufficiency and ischemia and culminate in the infarctions and demyelination that underlie the clinical signs and symptoms of CADASIL.
The structure of the granular osmiophilic material may hold clues regarding the pathogenesis of CADASIL. Lorenzi and coauthors conducted a study in which they examined the ultrastructure of the granular osmiophilic material in detail (73). They examined images of the granular osmiophilic material, generated through electron microscopy, and found that the granular osmiophilic material deposits in CADASIL are partially or totally surrounded by an electron-lucent halo. This halo is absent in other diseases that also have granular osmiophilic material, such as glomerulonephritis and focal segmental glomerulosclerosis. Thus, the presence of the electron-lucent halo sets CADASIL apart from these other granular osmiophilic material-containing diseases and suggests that the halo may play a unique role in CADASIL pathology or pathogenesis. The authors hypothesize that the halo is the site of aberrant Notch3 processing (73).
The deposition of Notch3 ectodomain at the plasma membrane of vascular smooth muscle cells not only leads to deposits of granular osmiophilic material but also facilitates interactions with key components of the vascular extracellular matrix and promotes their accumulation within the deposits. In particular, TIMP3 (a member of the tissue inhibitor of metalloproteinases family) and vitronectin (an adhesive glycoprotein) are proteins that are predominantly associated with the extracellular matrix of vessels. Both of these proteins abnormally accumulate in the Notch3 ectodomain–containing deposits. However, until recently, it was unclear whether the increased presence of these particular proteins contributes to the pathogenesis of CADASIL or is a mere epiphenomenon. A study by Capone and colleagues, utilizing mouse models of CADASIL, demonstrated that the increased accumulation of TIMP3 impairs cerebral blood flow autoregulation, whereas the increased accumulation of vitronectin worsens white matter lesions (16). Thus, excess levels of extracellular matrix proteins contribute to the compromised cerebrovascular reactivity and leukodystrophy of CADASIL (16).
The arteriopathy of CADASIL manifests itself principally as ischemia. However, the arteriopathy can also induce hemorrhages. Reports have demonstrated that patients with CADASIL can have spontaneous intracerebral hemorrhages, whether on antiplatelet therapy, anticoagulant therapy, or neither (68; 106).
Although the disease manifests itself solely as brain dysfunction, the vasculopathy of CADASIL is systemic. Ultrastructural arterial lesions similar to those found in the brain have been identified in spinal cord, spleen, heart, muscle, skin, and sural nerve (37; 18).
The characteristic angiopathic changes in muscle, skin, and sural nerve can be detected by biopsy, verifying the diagnosis of CADASIL. However, most pathology in CADASIL is restricted to the brain.
A high prevalence of right-to-left shunt, probably due to patent foramen ovale, is present among patients with CADASIL (108). It was originally thought that the presence of a right-to-left shunt may worsen the course of disease. However, no significant clinical or radiographic differences have been found between CADASIL patients with and without a right-to-left shunt. The high prevalence of right-to-left shunt in CADASIL may be due to a role of Notch signaling in cardiovascular development. Indeed, Notch3 is expressed in heart precursors during embryogenesis. Notch mutations likely lead to abnormal development of the endocardial cushion (81).
CADASIL is an autosomal dominant disorder caused by mutations in Notch3 on chromosome 19q13 (48). Notch3 is a 2321 amino acid glycosylated transmembrane receptor that plays a role in specifying cell fate during development (107; 05). The Notch signaling pathway was first identified and examined in the fruit fly Drosophila melanogaster. The name "Notch" was derived from the phenotypic notched wing found in flies that possess only one functioning copy of the gene. In flies, homozygous mutations are lethal (06).
Notch signaling influences cell-fate decisions by regulating gene expression. When Notch interacts with one of its ligands on a neighboring cell, the cytoplasmic domain of Notch is released from the cell surface and translocates to the nucleus where it acts as a transcriptional activator. In the adult, Notch signaling is thought to be important in tissue homeostasis by controlling proliferation, differentiation, and cell survival (51).
In humans, there are four Notch homologues (Notch 1 to 4) and five known ligands (Jagged 1 and 2 and Delta-like 1, 3, and 4). The expression of Notch receptors and ligands is regulated by tissue and developmental stage. During early stages of mammalian embryonic development, Notch3 is expressed broadly. In contrast, in the adult human, Notch3 expression is restricted to vascular smooth muscle cells (57). Besides CADASIL, the only human disorder known to involve a Notch gene is an adult T-cell leukemia, which is associated with a truncation of the Notch1 transcript (36). Mutations in the ligand Jagged 1 cause Alagille syndrome, an autosomal dominant disorder characterized by developmental abnormalities of the liver, heart, eye, skeleton, and other organs. However, the CADASIL phenotype does not include any known developmental disorder or neoplasia.
Because CADASIL is autosomal dominantly inherited, only one copy of the mutated gene is necessary to cause disease. However, under some unusual circumstances, an individual may inherit two mutated copies. A study compared the phenotype of CADASIL in patients who were heterozygous versus homozygous for the mutated allele. The disease characteristics and MRI imaging features were similar for both genotypes but tended to be more severe for the homozygous patients. This finding suggests that there may be a gene dosage effect for the pathogenesis of CADASIL (02).
Although the pathogenesis of CADASIL remains unknown, research has begun to shed light on the molecular mechanisms by which the defective Notch3 protein produces the pathophysiologic changes of CADASIL (57). The extracellular ligand-binding domain of Notch3 contains 34 tandem epidermal growth factor-like repeats, each of which has six conserved cysteines. So far, all of the mutations leading to CADASIL have been found either to create or destroy a cysteine residue in the epidermal growth factor-like repeats of Notch3 (62). The loss or addition of a cysteine residue induces aberrant disulphide bridge formation and improper folding of Notch3. These conformational changes alter the structure and processing of the protein and lead to defects in ligand binding, signaling, or clearance of the Notch3 extracellular domain (37).
In the healthy adult brain, Notch3 expression is restricted to vascular smooth muscle cells. In patients with CADASIL, vascular smooth muscle cells degenerate as granular osmiophilic material is deposited around them. Several studies have begun to elucidate the relationship between Notch3 gene mutations and morphological deposition of granular osmiophilic material around vascular smooth muscle cells (54; 80). In the first of these studies, immunostaining of skin biopsies from patients with CADASIL revealed that the granular osmiophilic material around vascular smooth muscle cells was specifically labeled with antibodies against the extracellular portion of Notch3, but not with antibodies recognizing the intracellular Notch3 domain. Furthermore, in skin sections from patients without CADASIL, no antibody binding was detected around the small dermal arteries. These results suggest that the major component of granular osmiophilic material in patients with CADASIL is the ectodomain of the Notch3 gene product (54).
A second study has gone even further to link the Notch3 mutations to small vessel pathology (80). Mutations of Notch3 cause accumulation of the ectodomain of Notch3 to accumulate in disulphide cross-linked aggregates in small vessels and to sequester several proteins of the extracellular matrix within these aggregates. These extracellular matrix proteins include tissue inhibitor of metalloproteinases 3 (TIMP3) and vitronectin (VTN), which are proteins that do not normally associate with Notch3, in its healthy state. Furthermore, the functions of these proteins are altered when sequestered into aggregates, and their altered functions promote vessel fibrosis. Thus, it appears that the accumulation of the mutated Notch3 ectodomain plays a central role in CADASIL pathogenesis by recruiting and sequestering additional proteins and corrupting their functions (80).
In addition to the inability to effectively remove the abnormal Notch3 protein, patients with CADASIL may also have a defect in Notch signaling, although this has not yet been definitively demonstrated in vitro or in vivo. Biochemical experiments suggest that some CADASIL Notch3 mutations have decreased expression at the cell surface, ligand-binding capability, or signaling activity, whereas other CADASIL mutants have normal signaling in vitro (50; 61; 89). Differences in Notch3 signaling in CADASIL could be due to the level of glycosylation of mutated and misfolded epidermal growth factor-like repeats. Notch3 ligand binding and signaling are modulated by glycosyltransferases such as Fringe (49). Biochemical studies have shown that Fringe has impaired activity on Notch3 fragments bearing common CADASIL mutations. These hypo-glycosylated Notch3 fragments form aberrant dimers in vitro (04). Impaired glycosylation of Notch3 in patients with CADASIL could affect signaling or clearance of the extracellular domain. Even if the mutant Notch3 protein signals normally, the accumulated Notch3 extracellular domain may bind all available ligands without transmitting a signal and, thus, dominantly inhibit the Notch3 signaling pathway.
The creation of a powerful animal model for CADASIL holds great promise to elucidate the disease pathogenesis (56). Initial attempts to model CADASIL in the mouse met with limited success as “knock-in” models failed to express the mutant gene at adequate levels or in the proper distribution and consequently failed to produce either vascular lesions or brain pathology. However, using an artificial chromosome approach, scientists have over-expressed Notch3 in an endogenous-like pattern in mice and have reproduced many of the cardinal pathological features of the disease. In particular, these transgenic mice, harboring a CADASIL-causing mutation, develop early-onset pathognomonic Notch3 aggregates and granular osmiophilic material (GOM) deposits in brain vessels, slowly progressive degeneration of the white matter, and cerebral hypoperfusion. Thus, the animal model closely mimics many of the pathologic changes of humans with CADASIL. The only major pathologic change of CADASIL that the mouse model does not recapitulate is lacunar infarcts, perhaps due to the lower ratio of white matter to gray matter in mice than in humans, or perhaps due to the much shorter life span of mice than humans.
The animal model has already shed considerable light on several pathogenic issues in CADASIL. It has been known that reduced cerebral perfusion and loss of white matter coexist in CADASIL, but it was unclear which was cause and which was effect. The animal model has shown that reduced cerebral blood flow precedes the loss of white matter, thus, strongly suggesting that cerebral hypoperfusion causes the leukodystrophy and not the other way around (56).
A study utilizing the transgenic mice has shown that the CADASIL mutation increases infarct size following a transient filament middle cerebral artery occlusion. In addition, the study showed that the mutation increases the cerebral blood flow threshold below which infarction ensues, even when blood vessel anatomy and degree of collateralization are unchanged (83). This finding suggests that the mutation increases the sensitivity of brain tissue to ischemia. Indeed, the researchers found an enhanced susceptibility to spreading depression, which are intense pandepolarization waves that slowly propagate across brain tissue and are accompanied by loss of transmembrane ion gradients and a massive efflux of potassium from intracellular stores to the extracellular space. Although spreading depression requires substantial cellular energy to correct, it likely worsens the supply-demand mismatch in the ischemic penumbra, thus increasing infarct size. Thus, the mutation not only disturbs cerebral blood supply but also introduces physiologic changes that render the brain parenchyma more vulnerable to the damaging effects of that altered blood supply (83).
The animal model has also helped to clear up a longstanding question regarding the mechanism of disease in CADASIL. It has been known for many years that CADASIL is an autosomal dominant disease with toxic gain-of-function properties to the mutant Notch3. Nevertheless, the possibility existed that, in addition to the toxic gain of function, the mutation may also produce disease through loss of function, as a result of haploinsufficiency. To address this issue, Cognat and colleagues utilized several mouse strains with Notch3 mutations, including mice with constitutive or conditional reduction of Notch3 activity, mice harboring the Notch3 mutation at the endogenous Notch3 locus, and mice overexpressing the Notch3 mutation on either a wild type or null background (23). The investigators found three lines of evidence converging toward a single conclusion. First, the Notch3 mutation at the endogenous Notch3 locus does not mitigate Notch3 activity in brain arteries. Second, elimination of wild-type Notch3 in mice overexpressing the CADASIL-causing mutation of Notch3 did not exacerbate the onset and burden of CADASIL pathology. Third, Notch3 null mice did not develop any of the neuropathology of CADASIL. Thus, CADASIL can develop without impairment of Notch3 signaling and is not likely due to loss of function of Notch3. Instead, the results strongly suggest that the driving mechanism of CADASIL is a toxic gain of function.
An additional study utilizing multiple lines of transgenic mice, histopathology, imaging, and molecular and genetic approaches has further reinforced the conclusion that the disease is due to a toxic gain-of-function and not to aberrant NOTCH3 signaling (33). This study has further shown that there is a critical role for Notch3 extracellular domain accumulation in CADASIL arterial pathology through the coaggregation of both wild type and mutant NOTCH3. Thus, mutant and wild type Notch3 accumulation together is a major driver of the arterial smooth muscle cell loss in CADASIL, and this mixed-protein accumulation may constitute a major target for therapeutic strategies.
The animal model has also shed light on the pathogenesis of migraine headaches in CADASIL. Cortical spreading depression is the electrophysiological substrate of migraine aura. Research has shown that cortical spreading depression is enhanced in mice expressing a vascular Notch3 CADASIL mutation or a Notch3 knockout mutation (35). Furthermore, the phenotype was stronger in the knockout mice, suggesting that loss of function in Notch3 in the vasculature underlies the pathogenesis of migraine in CADASIL. It is hoped that this powerful animal model will prove useful, not only to elucidate the pathogenesis of CADASIL but also to develop therapies for CADASIL and other small-vessel diseases of the brain (37; 18).
|
• Although CADASIL is rare, it is the most prevalent hereditary cerebral small-vessel disease. |
The prevalence of CADASIL is unknown. It was once thought to be an extremely rare disorder. However, since 1993, several hundred families with CADASIL have been identified, suggesting that the disorder is more common than previously recognized. Most of the affected families have been identified in Western Europe. Several families with CADASIL have also been reported from Japan and other Asian countries. The first North American family with CADASIL linked to chromosome 19 was reported in 1996 (10). Mutations in the Notch3 gene seem to be responsible for CADASIL in patients across ethnic groups. It is likely that the geographical differences in the reporting of CADASIL are a reflection of differences in disease prevalence (due to differences in population genetics) and not simply due to a lack of recognition.
As CADASIL is inherited in an autosomal dominant pattern, recognition of this disorder has relied heavily on the presence of a strong family history of similar symptoms. However, a Notch3 mutation underlying CADASIL can arise de novo as demonstrated by a patient with signs and symptoms strongly suggestive of CADASIL (migraine, stroke, and white matter abnormalities) but with no first-degree relative with similar signs or symptoms (59). This patient carried a heterozygous mutation in the Notch3 gene, but the mutation was absent in both of his biological parents. This finding demonstrated that CADASIL can be present in patients even in the absence of a family history.
Another finding has substantial implications regarding the prevalence of CADASIL. Pathogenic variants in the EGFr 7-34 of NOTCH3 have been found to be relatively prevalent in the general population and are much more prevalent than pathogenic variants in EGFr 1-6. Furthermore, the EGFr 7-34 mutations tend to produce a milder phenotype, later onset of disease, and higher survival rates than the EGFr 1-6 mutations (11). These findings suggest that CADASIL may be much more prevalent than previously appreciated and is likely responsible for a proportion of the large number of strokes, migraine headaches, and psychiatric complaints for which no definitive etiology is identified (95).
Currently, no known mechanism of preventing the symptoms and signs of CADASIL in individuals carrying the abnormal gene exists.
The most frequent initial symptom of CADASIL, stroke in middle adulthood, has a wide differential diagnosis. However, the combination of the age of onset, characteristic clinical course, occurrence in the absence of traditional vascular risk factors, neuroradiologic findings, laboratory profile, and inheritance pattern allows CADASIL to be differentiated from other disease processes in most cases. Genetic testing can verify the diagnosis.
The condition that most closely resembles CADASIL is Binswanger disease. Also known as "multiple subcortical infarction" and as "lacunar state," Binswanger disease is a condition of recurrent lacunar strokes, principally involving the subcortical white matter, resulting in progressive cognitive and motor decline. Neuroimaging generally shows multiple subcortical lacunes and a periventricular leukoencephalopathy sparing the arcuate subcortical U fibers. Pathologically, Binswanger disease manifests itself as small discrete infarcts and as larger areas of diffuse incomplete white matter infarction with extensive demyelination. The vascular lesion underlying the lacunes and leukoencephalopathy is a thickening and hyalinization of the media of small penetrating end-arteries and arterioles. Thus, the clinical course, neuroimaging, and pathological findings of Binswanger disease are similar to those of CADASIL (14). However, unlike Binswanger disease, CADASIL is not associated with hypertension or other vascular risk factors. Furthermore, CADASIL is a strongly familial disease, whereas Binswanger disease is not.
A second disease that may mimic CADASIL is multiple sclerosis. Although symptoms of CADASIL typically emerge during mid-adulthood, onset of CADASIL may occur decades earlier (at an age when multiple sclerosis presents as a common neurologic disease). The hallmarks of multiple sclerosis include a separation of symptoms in time and space and multiple white matter lesions on MRI. These symptoms are shared by CADASIL. However, neurologic symptoms in CADASIL commonly have a sudden onset, more suggestive of a stroke than of a typical exacerbation of multiple sclerosis. In addition, a strong family history is almost invariably present in CADASIL but often absent in multiple sclerosis.
Another disease that very closely resembles CADASIL, both clinically and by name, is CARASIL (cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy). CARASIL, inherited in an autosomal recessive pattern, typically has its onset in a person’s twenties or thirties and begins with spasticity of the legs and difficulty walking. Like CADASIL, CARASIL often causes strokes at young ages, mood and personality changes, and dementia. It differs from CADASIL by lacking a dominant pattern of inheritance. In addition, CARASIL has several features absent from CADASIL, including early hair loss from the scalp and back pain due to degeneration of spinal discs (82).
Like CADASIL, cerebral amyloid angiopathy may be associated with stroke, dementia, and leukoencephalopathy. Cerebral amyloid angiopathy usually presents in older patients than in those with CADASIL, but it may have an earlier onset in familial cases (41; 46). These conditions are distinguished by neuroimaging, which shows evidence of cerebral hemorrhage in cerebral amyloid angiopathy, but not in CADASIL.
Mitochondrial diseases may sometimes resemble CADASIL because of the progressive encephalopathy, recurrent strokes or stroke-like episodes, and white matter disease evident on MRI scans in some patients (01). Furthermore, like CADASIL, mitochondrial diseases may be familial. However, laboratory work up for mitochondrial disorders will typically reveal elevated serum lactate and pyruvate levels, ragged-red fibers on trichrome staining of muscle biopsies, abnormal mitochondrial enzyme activity in muscle, and defects in the mitochondrial genome. All of these laboratory abnormalities are absent in CADASIL. A study investigating the prevalence of the mitochondrial DNA mutation leading to MELAS (mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes) in patients with genetically unassigned CADASIL-like syndrome found the mutation in 0 out of 429 cases. This finding suggests that MELAS should be low on the differential diagnosis of patients with CADASIL-like symptoms (69).
Up to 30% of patients with CADASIL are initially diagnosed with complicated migraine headache, including migraine with visual or sensory aura, or with focal deficits (26). Migraine is usually diagnosed before the age of 40. Both familial migraine and CADASIL may present with severe headache, transient or permanent neurologic deficits, and a strong family history of a similar condition. Indeed, a locus for autosomal dominant hemiplegic migraine maps to the same region of chromosome 19 as CADASIL, and the two conditions were briefly hypothesized to be allelic (20; 21). Other evidence, however, has demonstrated that the two conditions are genetically distinct (28; 58). CADASIL can be distinguished from complicated migraine headache by the leukoencephalopathy evident on MRI in cases of CADASIL, but not in migraine headache. Furthermore, the clinical course is different in the two diseases, with pseudobulbar palsy and progressive dementia typically occurring in CADASIL, but not in migraine headache.
CADASIL is a heritable disorder that results in cerebral infarctions; therefore, its differential diagnosis may include a large group of familial diseases for which stroke may be a symptom. Such disorders include the hereditary dyslipoproteinemias (eg, familial hypercholesterolemia), heritable disorders of connective tissue (eg, Ehlers-Danlos type IV, Marfan syndrome, and homocystinuria), organic acidemias (eg, methylmalonic acidemia), mitochondrial encephalomyopathies (eg, MELAS and MERRF), and neurocutaneous syndromes (eg, neurofibromatosis and tuberous sclerosis). All of these disorders, however, can be distinguished from CADASIL by the characteristic findings on the general physical examination, by the pattern of inheritance, or by the laboratory abnormalities that define these disorders and that are absent in CADASIL.
Finally, recurrent strokes in relatively young patients may also be due to a hypercoagulable state, cerebral vasculitis, illicit drug use, or cardiac disease. However, patients with CADASIL typically have a normal or negative coagulation profile, cerebral angiogram, drug screen, echocardiogram, and ECG.
The mutated gene responsible for CADASIL (Notch3) has been identified and characterized, and genetic testing for the disease has become commercially available. Prior to development of genetic testing for CADASIL, the disease could be diagnosed only through a combination of clinical history, physical examination, and nonmolecular laboratory testing. Despite the availability of genetic testing, CADASIL can still be reliably diagnosed using clinical and nonmolecular criteria.
Clinical diagnosis. An important procedure to establish the diagnosis of CADASIL is MRI of the brain that will reveal evidence of multiple subcortical lacunar infarcts and a diffuse leukoencephalopathy. In many cases, these pathological changes will be evident to some degree on MRI years before the clinical onset of strokes or dementia. Therefore, MRI can potentially be used to diagnose CADASIL in at-risk presymptomatic individuals (63).
The presence of white matter hyperintensity on T2-weighted MRI images in temporal lobe poles is highly specific for CADASIL, in comparison to patients with hypertensive white matter changes (85). In patients with genetically determined CADASIL, white matter changes in the anterior temporal pole on MRI had a sensitivity of 89% and specificity of 86% for CADASIL. Involvement of the external capsule has a high sensitivity (93%) but is not as specific (45%) for CADASIL. In contrast, skin biopsy is only 45% sensitive but 100% specific for CADASIL (76).
For individuals with an established family history of CADASIL, a characteristically abnormal MRI is necessary and usually sufficient evidence to establish the diagnosis.
Patients presenting with symptoms of CADASIL and a family history of strokes, but no firmly established family history of CADASIL require a much more extensive diagnostic evaluation in order to assess for other causes of stroke (familial and otherwise). This laboratory evaluation should begin with the "routine" labs obtained for patients with stroke, including a complete blood count and white blood cell differential, coagulation profile, serum electrolytes, renal and liver function tests, blood glucose, platelet count, erythrocyte sedimentation rate, and lipid profile. All of these laboratory studies will typically be normal in patients with CADASIL. A duplex carotid ultrasound, ECG, and chest x-ray should be performed to assess for carotid and cardiac sources of embolism. The results of these procedures are all typically negative in patients with CADASIL.
Depending on the age, past medical history, and other items peculiar to each patient, a set of more specialized laboratory tests may be obtained, such as a serum amino acid analysis, VDRL, and antinuclear antibody to assess for homocystinuria, neurosyphilis, and lupus, respectively. Again, all of these laboratory tests would be expected to be negative or normal in CADASIL.
Pathologic diagnosis. Biopsy of skin, muscle, or sural nerve has been used to diagnose CADASIL, as arteries in the periphery develop ultrastructural changes similar to cerebral arteries. These pathognomonic abnormalities on biopsy can often be seen before the onset of strokes or dementia (34). Finding granular osmiophilic material on biopsy is specific for CADASIL, but the sensitivity of the angiopathic changes in peripheral tissues may be as low as 45% (76). The sensitivity of skin biopsy for CADASIL can be greatly increased by ensuring that a biopsy of sufficient depth and size is obtained. In particular, the skin biopsy should include the border zone between the deep dermis and the upper subcutis, where small arterial vessels of the correct size reside (101).
Some studies of families affected by CADASIL have found that skin and muscle biopsies are reliable in establishing the diagnosis (77) as both clinically affected subjects and clinically asymptomatic patients with minimal MRI abnormalities had characteristic abnormalities of granular electron-dense deposits. The discovery that CADASIL mutations induce an accumulation of Notch3 protein at the cytoplasmic membrane of vascular smooth muscle in the periphery as well as in the brain has led to the development of immunostaining techniques for the diagnosis of CADASIL (60). Skin biopsies are less invasive than muscle biopsies and less costly than a search for a mutation in the large Notch3 gene. Thus, electron microscopy and immunostaining of skin biopsy specimens has emerged as a powerful method in the diagnosis of CADASIL.
Molecular diagnosis. Molecular genetic testing for CADASIL has become available on a commercial basis. Two types of molecular genetic tests for CADASIL are available. The first type is linkage analysis for chromosome 19. This type of testing may be conducted appropriately for patients with multiple affected family members. The second type is direct sequencing of the Notch3 gene to identify point mutations in individuals. An important advantage of sequence analysis over linkage analysis is that the former allows individual patient testing without having to recruit family members or to rely on a dubious family history (48).
For the molecular diagnosis of CADASIL, sequencing of the Notch3 gene is undertaken in a stepwise strategy. The first step is to sequence exon 3 and 4, which is the site of the mutation in two thirds of cases. If no mutations are found in exons 3 and 4, the remaining exons that code for EGF-repeat-like domains should be sequenced. Together, the first two steps will identify the Notch3 mutation in 90% of CADASIL cases. Sequencing of the entire Notch3 gene is undertaken only if no mutation is found in the first two steps.
Genetic testing for CADASIL in all patients with lacunar strokes who have no other typical features of CADASIL has a low yield (29). Therefore, genetic testing should be initiated with patients who have the characteristic signs of lacunar stroke, white matter changes on MRI, family history (if available), and skin or nerve biopsy.
The nature of CADASIL and the inability of modern medicine to influence its course dictate that ethical considerations are addressed prior to genetic testing for the disease, especially in asymptomatic at-risk individuals. CADASIL has no known specific treatment and ultimately leads to severe disability, dementia, and death, thus, presymptomatic testing requires protocols similar to those used for Huntington disease (53). A study of at-risk people seeking presymptomatic genetic testing for CADASIL has demonstrated that most applicants choose not to complete the testing or to obtain their genetic status once the disease and the implications of genetic testing are fully explained to them (92).
|
• There is no definitive treatment of proven efficacy for CADASIL. | |
|
• Standard supportive treatment for strokes should be used. | |
|
• Anticoagulants, including intravenous thrombolysis and oral anticoagulants, should be used only with caution because of the increased risk of hemorrhage. | |
|
• Migraine and psychiatric disturbances should be treated symptomatically with standard treatments. |
Most patients with CADASIL are treated in a similar fashion to patients with lacunar strokes of other etiologies. This includes the use of antiplatelet medications. There is no evidence that any form of treatment changes the natural history of CADASIL. However, one study has shown significant correlations between the number of cerebral microhemorrhages, elevated systolic blood pressure, and hemoglobin A1c levels in patients with CADASIL (105). These results suggest that modulation of blood pressure and glucose levels could influence the course of CADASIL. Case reports describing intracerebral hemorrhages have led some authors to suggest that antiplatelet and anticoagulant medications should be avoided in patients with CADASIL (68).
Patients with CADASIL have an increased risk of microbleeds and overt intracranial hemorrhage. This increased risk of hemorrhage is thought to be due, at least in part, to loss of autoregulation. Because of this increased risk of hemorrhage, some have discouraged the use of intravenous tissue plasminogen activator (IV tPA) for CADASIL patients with an acute stroke. However, there is growing evidence to support the use of tPA in ischemic stroke associated with CADASIL. A retrospective analysis of case studies suggests that tPA may actually lower the risk of hemorrhagic complications, which are frequent in CADASIL (43). Thus, IV tPA should be considered as a treatment option and should not be considered contraindicated in patients with CADASIL (66).
Cholinesterase inhibitors can improve cognition, activities of daily living, and global functioning in patients with mild to moderate Alzheimer disease. Enhanced cholinergic signaling has this beneficial effect in Alzheimer disease because cholinergic input from the basal forebrain to the cerebral cortex is lost as part of Alzheimer disease pathophysiology. Likewise, in CADASIL, it is likely that cholinergic pathways from the basal forebrain to the cortex are disrupted by subcortical ischemic lesions. Thus, a clinical trial was conducted to determine whether donepezil, a potent cholinesterase inhibitor, can improve cognition in patients with CADASIL (27). The authors found that donepezil did not improve cognition, the study’s primary endpoint. However, donepezil did appear to improve several measures of executive function and processing speed, the study’s secondary endpoints. The beneficial effect of donepezil on executive function in CADASIL likely reflects the disruption of cholinergic fibers to the dorsolateral prefrontal cortex, a projection known to be disrupted by subcortical strokes in CADASIL. Although this single study showed a positive effect of donepezil on several secondary outcome measures in CADASIL, regulatory approval for the use of cholinesterase inhibitors in CADASIL must await further clinical trials.
Nitric oxide is synthesized in the endothelium. Increased nitric oxide concentrations at the endothelium preserve cerebral blood flow and prevent inflammation, platelet aggregation, thrombosis, and apoptosis. Tetrahydrobiopterin is an essential cofactor for nitric oxide synthesis in endothelial cells. Therefore, De Maria and colleagues examined whether supplementation with sapropterin, a synthetic tetrahydrobiopterin analog, could improve endothelium-dependent vasodilation in patients with CADASIL (25). They conducted a 24-month, multicenter, randomized, double-blind, placebo-controlled trial in which patients with CADASIL were randomly assigned to receive placebo or sapropterin. The primary end point was change in the reactive hyperemia index by peripheral artery tonometry at 24 months. Safety and tolerability of sapropterin were also assessed. The researchers found that sapropterin was safe and well-tolerated but did not affect endothelium-dependent vasodilation in patients with CADASIL. Thus, sapropterin will likely not be a useful treatment for CADASIL.
Because migraine with aura is often a prominent symptom, especially in the early stages of CADASIL, identification of an effective prophylaxis against this component of the disease would be useful, even if the infarcts and leukoencephalopathy cannot ultimately be prevented. Multiple anecdotal reports and one retrospective analysis have suggested that acetazolamide may be particularly effective in prophylaxis against migraine with aura in CADASIL (30).
Drug information sheets often state that triptans are contraindicated in patients with stroke or transient ischemic attack because of the theoretical risk that they could trigger or worsen cerebral ischemia. For this reason, some have argued that triptans should not be used to treat acute migraine headaches in patients with CADASIL because they are already at increased risk for stroke. However, a large study examining migraine patterns and treatment responses in CADASIL found that the migraine headaches of CADASIL tended to respond well to the triptans and that no complications were induced. Thus, triptans can be used safely and effectively to treat migraine headaches in patients with CADASIL (100).
Although no clinical study has yet revealed an effective treatment for CADASIL, several preclinical studies, both in vitro and in vivo, have yielded promising results. Stem cell factor and granulocyte colony-stimulating factor are hematopoietic growth factors that play critical roles in regulating blood cell production, as well as bone marrow stem cell survival and mobilization. Their physiologic role in the central nervous system is unknown, but studies have shown that these two proteins are neuroprotective and reparative in animal models of ischemic stroke. Therefore, Liu and associates examined the possible therapeutic effects of stem cell factor in combination with granulocyte colony-stimulating factor in a mouse model of CADASIL (72). They found that stem cell factor plus granulocyte colony-stimulating factor attenuated vascular smooth muscle cell degeneration in small arteries, increased cerebral vascular density, and inhibited apoptosis in the CADASIL mice. The treatments also led to functional improvement, as they improved cognitive function on the water maze. Thus, the combination of stem cell factor and granulocyte colony-stimulating factor restricts pathological progression and improves the outcome in CADASIL, at least in mouse models of the disease.
A separate preclinical study examined the possibility that correction of Notch3 function could abrogate the pathology of CADASIL. Toward this end, Machuca-Parra and colleagues administered an agonist Notch3 antibody (75). They showed that the antibody activates a ligand-insensitive mutant Notch3 receptor in vitro and that it prevents mural cell loss in vivo in mice with the CADASIL mutation. These results suggest that CADASIL could ultimately be treated successfully through the use of agents that modulate Notch3 signaling.
Another novel antibody was developed against the Notch3 ECD. In mutant mice inoculated with this immunotherapy, the aggregation of NOTCH3 within both the vasculature and the serum significantly decreased (84). In theory, lower densities of the Notch3 ECD aggregations will reduce the risk of lacunar infarcts and, therefore, illness severity in CADASIL. Though there have not yet been any immunomodulatory therapies developed for humans, such antibodies against the Notch3 receptor are promising for human use.
A separate approach to immunotherapy for CADASIL would be to utilize antibodies directed against the granular osmiophilic material, which is the substance that impairs blood flow and leads to strokes. Granular osmiophilic material is the accumulation of the extracellular domain of the NOTCH3 receptor. Thus, it follows that administration of an antibody directed against the NOTCH3 extracellular domain could lead to clearance of the granular osmiophilic material and improved outcomes. In a preclinical study utilizing mutant mice that overexpress NOTCH3 and produce the pathology of CADASIL, Ghezali and colleagues administered repeated injections of antibodies directed against the extracellular domain of Notch3 (38). They found that the antibodies strongly and specifically adhered to granular osmiophilic material but did not attenuate granular osmiophilic material deposition or halt the development of white matter lesions. However, the antibody treatments did protect against impaired cerebral blood flow and normalized myogenic responses of cerebral arteries. Thus, immunotherapy targeting with antibodies targeting the NOTCH3 extracellular domain could become a disease-modifying treatment for CADASIL (38).
Rather than developing an entirely new drug or antibody, some researchers have been investigating the use of existing medications to potentially treat or prevent CADASIL. One study in mice has found that cerebrolysin, a neuropeptide used in dementia and Alzheimer disease treatment, is associated with increased longevity (65). Thus, although the trials with cerebrolysin have so far been conducted only in mice, the results demonstrate the principle that existing drugs or therapies might be retooled for people with CADASIL.
Although CADASIL is a genetic disorder, the ultimate goal is to cure the disease using gene therapy. An important first step toward this goal has been taken. As described earlier, the CADASIL mutation alters the number of cysteine residues in one of the epidermal growth factor repeat domains from an even number to an odd number of cysteines. This leads to an unpaired cysteine residue and disrupted disulphide bridge formation in the mutant Notch 3 extracellular domain. This, in turn, leads to increased multimerization, which induces toxic aggregation of the protein in and around vascular smooth muscle cells. Although the uneven number of cysteines underlies the pathologic process, Rutten and colleagues reasoned that exclusion of the mutant epidermal growth factor repeat domain would abolish the detrimental effect of the unpaired cysteine and prevent the toxic accumulation of the protein (94). Utilizing the molecular approach of exon skipping, they showed that the modified protein can be expressed, can undergo normal protein processing, and can retain its ligand-binding activity and ligand-induced activation. The investigators next transfected antisense oligonucleotides into cerebral vascular smooth muscle cells derived from patients with CADASIL and showed that successful exon skipping could be achieved without diminishing NOTCH3 signaling. These findings demonstrate that rational molecular gene therapeutic approaches are potentially useful for CADASIL. However, to date, these approaches have only been applied in vitro. If methods can be devised to similarly alter the mutant protein in vivo, while avoiding unwanted off-target effects, then real change may lie ahead for patients with CADASIL.
One study has suggested strong and clinically important relationships between CADASIL and pregnancy (93). The frequency of neurologic symptoms and signs during pregnancy and the puerperium were studied in a cohort of 25 women with genetically confirmed CADASIL. Almost half of these women (12 of 25) experienced neurologic symptoms during gestation or the puerperium during at least one of their pregnancies. The most common of these neurologic signs and symptoms were hemiparesthesia, hemiparesis, aphasia, and visual disturbances. Confusion, vertigo, and dysarthria also occurred. In the great majority of these affected patients (82%), these neurologic signs and symptoms were the first clinical manifestation of CADASIL.
Preeclampsia was also more frequent (10.3%) in the patients with CADASIL than in the normal population (3% to 5%). Furthermore, in contrast to the normal population where preeclampsia occurs most commonly during the first pregnancy, in the patients with CADASIL, preeclampsia occurred most often in later pregnancies. Thus, pregnancy is a period during which women with CADASIL may have their first neurologic manifestations. In addition, CADASIL may be a risk factor for preeclampsia. CADASIL should, therefore, be considered when searching for the cause of transient or permanent stroke-like events during pregnancy or the puerperium.
A later study examining CADASIL in pregnancy found much more reassuring outcomes. In a study of 50 pregnant women with CADASIL, Donnini and colleagues found that none of the women had disease onset or suffered from cerebrovascular ischemic events during the pregnancy (31). They further found that delivery complication rates were not increased and that fetal growth and newborn weights were normal. These researchers concluded that CADASIL is not associated with unfavorable pregnancy outcomes for either the women or their fetuses. In addition, these researchers concluded that there is no need for specific antithrombic treatment during pregnancy in CADASIL.
The clinical manifestations linking CADASIL and pregnancy may be psychiatric as well as neurologic. During the puerperium, women with CADASIL may present with acute psychosis, and this may be their first sign of CADASIL (87). Thus, CADASIL should be considered in the differential diagnosis of women with postpartum psychiatric disturbances.
Another relevant point concerning the joint topics of pregnancy and CADASIL is that the condition may be diagnosable by MRI or by genetic testing in an at-risk presymptomatic pregnant woman or prospective father; thus, providing some important information for genetic counseling. A final important point regarding pregnancy and CADASIL is that prenatal testing for the disease is possible for fetuses using the same DNA-based techniques used for adults. DNA extracted from fetal cells obtained by amniocentesis or by chorionic villus sampling can be tested for mutations in Notch3 (79). Similarly, preimplantation genetic testing of embryos following in vitro fertilization can be utilized to select CADASIL-free embryos for implantation (67). In all cases, genetic counseling should precede molecular analysis. The same ethical issues regarding prenatal testing for Huntington disease pertain to CADASIL (53).
Acute lacunar infarcts following episodes of hypotension or surgery have been the presenting sign of CADASIL in some patients. These observations likely reflect a reduced capacity of the microvasculature to cope with hypotension in CADASIL. For this reason, it is essential to monitor blood pressure continuously during general anesthesia. It has been further emphasized that normocapnia and normothermia need to be maintained during surgical procedures for patients with CADASIL (09).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.
Marco Garcia MD
Dr. Garcia of Levine Children's Hospital has no relevant financial relationships to disclose.
See Profile
Daniel J Bonthius MD PhD
Dr. Bonthius of Atrium Health/Levine Children's Hospital has no relevant financial relationships to disclose.
See Profile
Steven R Levine MD
Dr. Levine of the SUNY Health Science Center at Brooklyn has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Sleep Disorders
Jul. 05, 2026
General Child Neurology
Jun. 24, 2026
General Child Neurology
Jun. 10, 2026
Epilepsy & Seizures
Jun. 02, 2026
Neurogenetic Disorders
Jun. 01, 2026
General Neurology
May. 13, 2026
General Child Neurology
May. 12, 2026
Neurogenetic Disorders
May. 08, 2026