










Pyruvate dehydrogenase complex deficiency
May. 28, 2024
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
This article includes discussion of childhood ataxia with central nervous system hypomyelination, CACH, myelinopathia centralis diffusa, and vanishing white matter disease,. The foregoing terms may include synonyms, similar disorders, variations in usage, and abbreviations.
Mutations affecting the eukaryotic initiation factor 2B (eIF2B) cause one of the most common leukodystrophies, the autosomal recessive childhood ataxia with central nervous system hypomyelination (CACH), or vanishing white matter disease (VWM). Patients may develop a wide spectrum of neurologic abnormalities, from prenatal-onset white matter disease to juvenile- or adult-onset ataxia and dementia, sometimes with ovarian insufficiency. The pattern of diffuse white matter abnormalities on brain MRI and diffusion studies is often diagnostic. A knock-in mouse model of CACH/VWM, which shows a developmental white matter abnormality, is a promising new tool for the research of this devastating disease. Molecules that stabilize the eIF2B complex and normalize its activity are being developed. Symptomatic treatment such as deep brain stimulation may alleviate tremor.
• Childhood ataxia with CNS hypomyelination (or vanishing white matter disease) is a relatively common leukodystrophy in which most of the patients have a pathognomonic pattern of MRI and diffusion tensor imaging abnormalities. | |
• Patients with childhood ataxia with CNS hypomyelination have a usual susceptibility to mild head trauma, fever, and other stresses. | |
• Childhood ataxia with CNS hypomyelination can present at any age. A detailed quantitative natural history study has been published. | |
• eIF2B mutations are associated with likely mitochondrial dysfunction that affects astrocytes and oligodendrocytes. | |
• Molecules like ISRIB (integrated stress response inhibitor) correct the eIF2B deficiency in most mutants and are likely to be tried as therapy for central nervous system hypomyelination/vanishing white matter disease. |
Patients with childhood ataxia with central nervous system hypomyelination were first identified in 1992 (53). Similar patients were soon described (25; 62). This disorder is also known as "the vanishing white matter disease" (62; 63).
Initially, some patients develop normally, whereas others have speech and cognitive delay (25; 52; 62). New-onset ataxia is the most common initial sign between the ages of 1 and 5 years (51). Some patients develop coma or dysmetric tremor. These can be spontaneous or follow a mild head trauma or febrile illness and even acute fright (78). Subsequently, deterioration is generally progressive with increasing difficulty walking, tremor, spasticity with hyperreflexia, dysarthria, and seizures. At any stage of the illness, patients may remain stable for years. Head circumference has been normal in all our patients. Swallowing difficulties and optic atrophy develop late in the disease, although the peripheral nervous system is unaffected (25; 52; 62). Death typically occurs during the first or second decade of life. Some patients develop symptoms after 5 years of age with a more slowly progressive spastic diplegia, a relative sparing of cognitive ability, and a likely long-term survival (52; 63). A rapidly fatal infantile form of this syndrome was described; it was found to be allelic to the more common form of the disease with the 3q27 locus (20). We found that a subacute variant of childhood ataxia with central nervous system hypomyelination occurs in the Cree indigenous population of Northern Canada (03; 19). These patients have an onset of neurologic deterioration in the first 6 months of life and die by 2 years of age. Congenital forms of the disease exist with manifestations in organs besides the brain (68). On the other end of the clinical spectrum are patients with a slowly progressive neurologic disorder, with onset after 5 years of age and that is often associated with ovarian insufficiency, which we called ovarioleukodystrophy syndrome (Schiffmann et al 1997; 16). Adult-onset disease is not rare and is probably underdiagnosed, particularly with the most common p.R113H EI2B5 mutation (17; 65). A series of 16 adult patients demonstrated the common presence of behavioral and psychiatric manifestations, as well as progressive cerebral atrophy, in addition to the more typical white matter findings on MRI (33). Adult-onset patients may present with isolated optic neuropathy (02). Cerebrospinal fluid protein and lactate, in addition to laboratory tests for the other known leukodystrophies, are normal. Although the majority of patients have been sporadic, 35% of our childhood ataxia with central nervous system hypomyelination families have had 2 siblings affected. Childhood ataxia with central nervous system hypomyelination is an autosomal recessive leukodystrophy syndrome; affected individuals are equally common in both sexes, and obligate carriers are asymptomatic (52; 62; 35).
Childhood ataxia with central nervous system hypomyelination is usually progressive (24). A large retrospective natural history study showed that the first disease signs occurred at a median age of 3 years; 60% of patients were symptomatic before the age of 4 years (24). Motor deficits were the most common presenting sign, especially in children. Adolescent and adult onset patients were more likely to exhibit cognitive problems early after disease onset. One hundred and two patients died. Absence of stress-provoked episodes and absence of seizures predicted more favorable outcome. In patients with onset before 4 years of age, earlier onset was associated with more severe disability and higher mortality. In patients with onset after 4 years of age, disease course was generally milder, with a wide variation in severity (24). Brainstem dysfunction often leads to breathing difficulties and death, mostly in the early-onset type of childhood ataxia with central nervous system hypomyelination, such as in Cree leukoencephalopathy. Most patients remain stable for years (52; 63). Disease severity is significantly correlated with age of onset (24). Some mutations in the homozygous state are clearly associated with either a mild or a severe form of the disease (17). Seizures are frequent in the early onset group late in the course of the illness. These are usually easy to control with antiepileptic medications.
A 7.5-year-old boy was admitted for evaluation of a leukodystrophy and an ataxic gait. He was well until 4 years of age, when he developed subacute onset of gait instability and "foot drop." The difficulties became worse, but a few weeks later he progressively improved, recovering an almost normal gait. At the age of 5, he fell and had a mild head trauma and could not stand independently. He had a slow recovery in the subsequent months and required a walker to assist in ambulation. A right ankle foot orthosis was used to facilitate ambulation. Fine motor movements were also affected; he had difficulties opening a door using a key. In the subsequent years, the patient remained mostly stable, showing only mild motor deterioration. His cognitive abilities were normal, and he attended a regular school and performed at grade level.
Family history was unrevealing; the patient was an only child. His examination revealed a head circumference in the 50th percentile, weight at the 80th percentile, and height above the 95th percentile. General examination was unremarkable. His speech was slow. Extraocular movements were normal, as was funduscopic examination. Mild tongue weakness was the only cranial nerve abnormality. He had moderate spastic weakness of the lower extremities in an upper motor neuron pattern. Fine motor movements of the upper extremities were markedly dysmetric. He could not stand unaided, but could make a few steps when supported by another person. Tendon reflexes were increased with ankle clonus and Babinski signs bilaterally. Sensory examination was normal to light touch, vibration, and temperature.
A nerve conduction study and electromyography were normal. MRI of the head showed diffuse increase in signal intensity on T2-weighted imaging and correspondingly decreased signal intensity in T1-weighted images.
Magnetic resonance spectroscopic imaging showed reduced choline-containing compounds, N-acetyl aspartate, and creatine in white matter only. An extensive laboratory evaluation consisting of routine hematologic, liver, and renal functions were normal as were tests for known leukodystrophies, lysosomal storage diseases, aminoacidopathies, and organic aciduria. Spinal fluid had normal glucose, lactate, and protein levels. Muscle biopsy was morphologically normal. Mitochondrial enzymes in muscle were normal; nerve biopsy was normal.
At this point, the clinical and neuroradiologic picture coupled with the unremarkable laboratory evaluation was consistent with childhood ataxia with central nervous system hypomyelination.
Genetic linkage to a region at chromosome 3q27 was found in a Dutch subgroup of childhood ataxia with central nervous system hypomyelination patients (34). The gene in this region was subsequently identified as EIF2B5, which codes for the epsilon subunit of the protein translation initiation factor eIF2B (35). It soon became clear, however, that this disorder is genetically heterogeneous and that it can be caused by mutations in any of the 5 subunits of eIF2B (64; 17; 51). Most patients are compound heterozygotes with a missense mutation in at least 1 allele (51). A rare submicroscopic deletion of 14q24.3 contributed to the disease in 1 patient (55). EIF2B-related disorder is pan-ethnic and has been described in a number of Chinese patients (81). Currently more than 120 mutations have been described in patients, with mutations affecting any of the 5 subunits of eIF2B (39). There are at least 2 mutations with known founder effect. These are the histidine substitution at arginine 195 of epsilon-eIF2B, R195H, and the A87V mutation in exon 3 of EIF2B3 in Quebec (19; 46). There is a significant degree of genotype-phenotype association for specific mutations. In particular, R113H and E213G in their homozygous and even compound heterozygous state are associated with a late disease onset and slow progression whereas V309L is associated with early onset and severe disease (17; 71). eIF2B is the guanine nucleotide-exchange factor for eIF2 and plays a key role in protein synthesis. Guanine nucleotide-exchange factor activity is reduced in immortalized lymphoblasts in correlation with disease severity in lymphoblasts (17b), but not in other cellular systems (31; 38).
The presence of the unique foamy oligodendrocytes, not seen in any other leukodystrophy, suggests that childhood ataxia with central nervous system hypomyelination represents a clinicopathologically homogeneous entity. Biochemical analysis of brain material confirms the impression of hypomyelination, but the abnormalities are nonspecific. Gross examination of brain sections reveals normal consistency of the cortical gray matter in marked contrast to the white matter of the centrum semiovale that is softened, atrophic, and gelatinous.
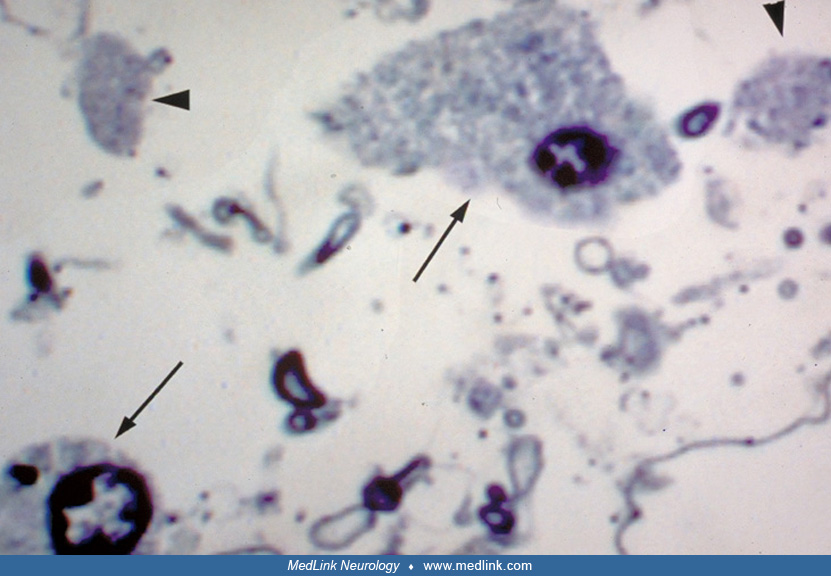
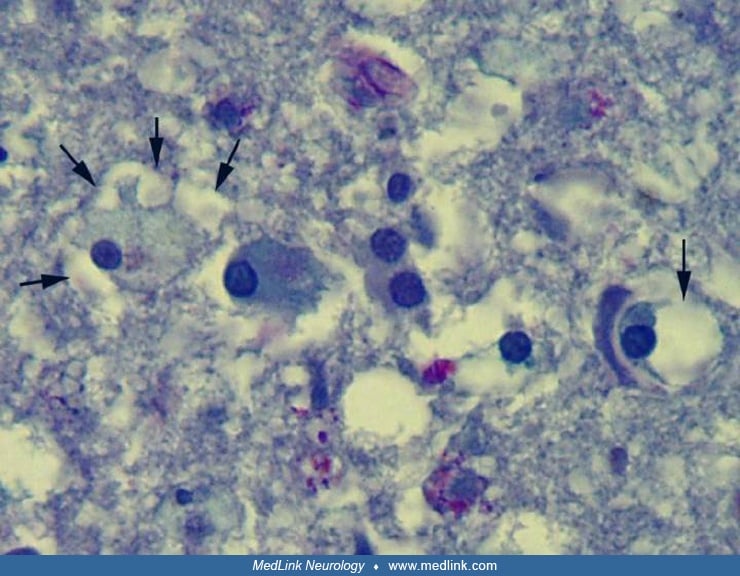
Light microscopy demonstrates rarefaction of the white matter with relative sparing of axons and subcortical U-fibers (52; 62). There is moderate to severe vacuolation of the white matter, scattered gliotic astrocytes, and rare to moderate numbers of macrophages. Increased numbers of oligodendrocytes were described by at least 2 groups (47; 75). Although caution is warranted in interpreting this as an indication of an actual increase in oligodendrocyte number because cell loss in the central nervous system produces an initial compaction of brain parenchyma, resulting in spurious increases of cellularity, this finding may be real (58; 49). Abnormal oligodendrocyte-like cells were identified by light and electron microscopic examination.
These cells have an enlarged cytoplasm with an eccentric nucleus 8µm to greater than 45µm in diameter.
These cells predominate in the deep white matter and are stained in a diffuse manner for various myelin proteins (oligodendrocyte glycoprotein, myelin basic protein, 2', 3' cyclic nucleotide 3' phosphodiesterase, and proteolipid protein) as well as histochemically for Luxol fast blue stain, but negative for macrophage markers.
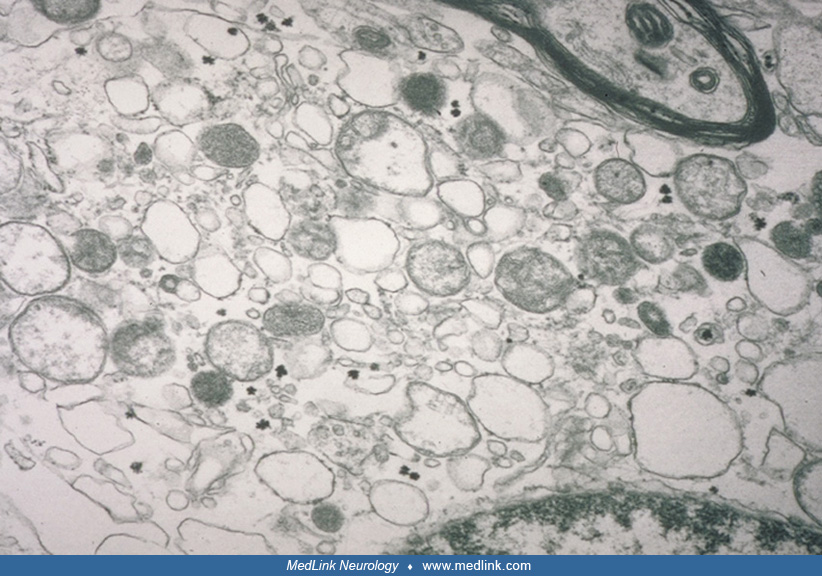
On electron microscopy, these oligodendrocytes look foamy with numerous round membranous inclusions that seem to come off the axonal sheath and mitochondria.
They clearly differ from macrophages with myelin-filled phagolysosomes (79). Similar cells were not seen in age-matched normal controls that were fixed and analyzed in the same manner or in several disease controls. Abnormal oligodendrocytes were also observed by other investigators (47).
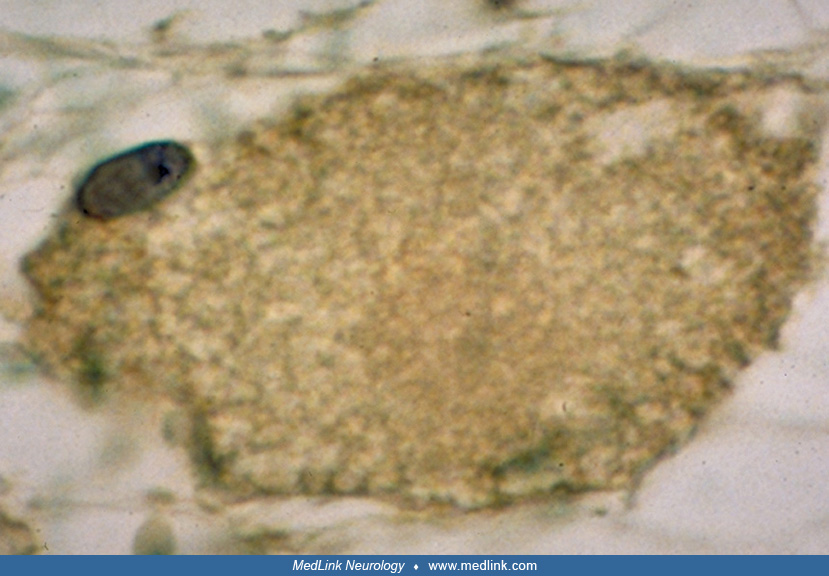
Based on the histologic criteria, childhood ataxia with central nervous system hypomyelination appears to be a predominantly glia-related disorder with relative preservation of axons noted in all 6 pathologically studied cases of childhood ataxia with central nervous system hypomyelination that we examined (43). Glial fibrillary acidic protein-positive glial fibers were seen aligning capillaries, and myelin sheaths around many of the axons were split, giving the impression of hypomyelination. Sural nerve biopsies on 4 of our patients were normal. Coarse, atypical astrocytes are also commonly found in these brains (79). In addition, a reduced number of astrocytes was found both in situ and in cultured patient astrocytes (20; 11). These astrocytes are thought to be immature, showing markers of early and late cellular differentiation (07). Interestingly, these same authors have found that the white matter of patients with CACH/VWM is enriched in CD44-expressing astrocyte precursor cells and accumulates the glycosaminoglycan hyaluronan (06). These findings may explain the positive Alcian blue/PAS staining in brain of these patients (Figure 14). Sural nerve biopsies on 4 of our patients were normal, but in 1 patient it was abnormal (50). It is likely that the abnormalities in oligodendrocytes and astrocytes seen in this syndrome point to a poor control of stress-related mechanisms of protein translation (35). Remarkably, inappropriate activation of the unfolded protein response was found in oligodendrocytes and astrocytes in white matter of childhood ataxia with central hypomyelination brains, whereas it was normal in the same cell types in the patients’ cortex and in the entire brains of normal controls (77). We have also found that the abnormal oligodendrocytes of children with childhood ataxia with central nervous system hypomyelination stain positively with Alcian blue.
This may be related to the glycosaminoglycan hyaluronan accumulation (06).
Immunohistochemical staining for major myelin proteins demonstrated the presence of these proteins in subcortical white matter (52). The Western blots from each of these myelin proteins revealed bands of expected size but reduced abundance compared to controls (52). The lipid extracts from childhood ataxia with central nervous system hypomyelination-ventricular wall motion brains were also reduced in galactocerebroside, sulfatide, and ethanolamine plasmalogen, although the ganglioside composition was normal (52). Proton-decoupled phosphorous magnetic resonance spectroscopy suggest that the eIF2B defect in this disorder causes an abnormality of myelin membrane synthesis or myelin membrane transport in the in vivo childhood ataxia with central nervous system hypomyelination brain. The authors found a decrease in glycerophosphorylethanolamine and an increase in phosphorylethanolamine, which suggested a defect in plasmalogen metabolism (04). It would be important to determine whether this abnormality is directly related to the basic defect in childhood ataxia with central nervous system hypomyelination or is only a secondary finding.
The mutations affecting any of the 5 subunits of eIF2B cause a partial deficiency of the guanine exchange factor activity of this protein (18; 37; 45). The most common mechanism is the exclusion of the mutated subunit from the entire 5 subunit complex. eIF2B, together with eIF2 and other proteins form a complex that has a major role in the regulation of protein translation. This complex is highly conserved and has an important role in preventing the accumulation of misfolded or denatured proteins during various stress conditions such as fever, infections, and, possibly, mechanical trauma (30). The mild reduction in basal activity of eIF2B activity may not be sufficient to explain the disease. There may be abnormalities associated with eIF2B function under physiological stress conditions. For example, decreased eIF2 phosphorylation in response to heat stress in lymphoblasts of eIF2B-mutated patients compared to controls was described (76). Although it is possible that affected oligodendrocytes end up dying by apoptosis (05), it is likely that it is the end result of a much more complex process. The reason for the selective involvement of glial cells in patients with eIF2B mutations is likely to be related to the unbalanced stoichiometry of subunits within large protein complexes that plays an important role, for example, in the mitochondria, which were found to be dysfunctional in astrocytes and oligodendrocytes (14; 27). Cultured skin fibroblasts expressing mutant EIF2B genes respond normally to stress conditions by reduced global translation rates, but they exhibit significantly greater increase in ATF4 induction compared to normal controls (31). The same author demonstrated a continuous hyper-stress state in oligodendroglial-derived cells (32). We think that glial cells are particularly susceptible to stress. The translation level of mRNAs of other genes, yet to be discovered, is abnormal under stress conditions in addition to the known ATF4 mRNA (50). The blend of induced transcription factors (ATF4 included) may have a major influence on transcriptional activity of key target genes, leading to direct or indirect effect on myelination and other glial cell functions. It seems that important factors leading to the disease in addition to EIF2B mutations are intrinsic to each cell type according to its nature. These parameters include the basal level of eIF2B activity and the stress level each cell type normally experiences. In support of the above concept is the active unfolded protein response state, which was demonstrated in glia of patients with childhood ataxia with central nervous system hypomyelination (74; 77). The ovarian insufficiency that is often seen in women with childhood ataxia with central nervous system hypomyelination (ovarioleukodystrophy) suggests that the function of eIF2B is also important for the development of ovarian cells. Interestingly, polymorphisms of EIF2B5 have been found to be significant prognostic factors in ovarian cancer (23).
One knock-in mouse model has been described that is homozygous for the R132H replacement corresponding to the clinically significant human R136H mutation of the EIF2B5 gene (22). The mouse exhibited hypomyelination similar to the one observed in the human disease and a delay in recovery from cuprizone-induced demyelination, reflecting an increased sensitivity to brain insults (22). These abnormalities are associated with unique time point-specific gene expression signatures indicating that EIF2B-mutated brains are forced to employ extra protective means during stressful periods, such as times of differentiation, synaptogenesis, and massive myelination (40). Furthermore, a mild reduction in eIF2B activity was associated with failure to adequately synthesize and secrete cytokines in response to lipopolysaccharide (LPS) treatment despite proper induction of cytokine mRNAs (08). Therefore, this mouse model prevents the appropriate increase in translation rates upon exposure to the inflammatory stressor LPS. These data suggest a downstream mechanism for the disease and underscore the importance of fully functional translation machinery for efficient cerebral inflammatory response upon insults (08). Using the mouse, investigators found unbalanced stoichiometry of proteins involved in oxidative phosphorylation and the mitochondrial translation machinery components (44). Mouse embryonic fibroblasts showed 55% decrease in oxygen consumption rate per mitochondrial DNA content and 47% increase in mitochondrial abundance (p < 0.005). Increased mitochondrial content was indeed found in patients’ oligodendrocytes (79). Primary mutated astrocytes, which are centrally involved in the disease, showed more severe eIF2B-associated oxidative respiration deficiency, with a 2-fold increase in basal and stimulated glycolysis (44).
Two more severe mouse models were generated: Eif2b5Arg191His/Arg191His and Eif2b4Arg484Trp/Arg484Trp (13). Astrocyte exhibited a maturation defect with associated secretion of factors that inhibit the growth of oligodendrocytes in culture (13). The defect in translation control is likely to lead to a mitochondrial dysfunction (21).
Major pathologic features | |
• Foamy oligodendroglial cell and possibly increase in the number of oligodendrocytes | |
• Rarefaction of the white matter (hematoxylin and eosin, Luxol fast blue) | |
• Relative axonal preservation compared to white matter loss | |
• Relative sparing of the white matter U-fibers | |
• Atypical perivascular astrogliosis with blunted processes and, in some cases, decrease in the number of astrocytes that are immature | |
Minor pathologic features | |
• Microgliosis, focal, absent to moderate in severity | |
• Macrophages, focal, absent to moderate in severity | |
The incidence of childhood ataxia with central nervous system hypomyelination is unknown. Although it is a rare disease, in our experience about half of the patients with previously unclassified leukodystrophies have been determined to have childhood ataxia with central nervous system hypomyelination. We estimate that more than 200 patients with childhood ataxia with central nervous system hypomyelination have been identified to date. Patients with CACH/VWM consisted of 5% of patients with leukodystrophies in the UK (56).
Prenatal diagnosis when the mutations affecting an EIF2B subunit are known is currently possible.
It should be stressed that childhood ataxia with central nervous system hypomyelination can be diagnosed with a high degree of confidence based on the pattern of abnormalities on the brain MRI. Therefore, the differential diagnosis should be mostly limited to other disorders with a similar assortment of MRI changes. These include mitochondrial disorders (10), leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation (48), and to a lesser extent adrenoleukodystrophy, Canavan disease, or certain forms of congenital muscular dystrophy (67). Multiple sclerosis may be confused with the adult-onset form of CACH/VWM (28). Spinal cord atrophy and post-contrast enhancement with enlargement of cranial nerves has been described (Eluvathingal et al 2018). Indeed, astrocytes were also found to be abnormal in the spinal cord of the mouse model and patients (36). When ovarian insufficiency occurs (ovarioleukodystrophy), the other main disorder to consider is AARS2-related ovarioleukodystrophy (61).
The initial diagnosis of childhood ataxia with central nervous system hypomyelination is clinical at present. It is mainly based on recognizing the typical changes on brain MRI (54). Once suspected, the diagnosis can be confirmed by the identification of mutations affecting any of the 5 subunits of eIF2B, possibly preceded by determination of asialotransferrin to total transferrin ratio in CSF (72). However, the latter does not exist as a clinical test yet. The cranial CT shows diffuse and symmetric hypodensity of the hemispheric white matter (52).
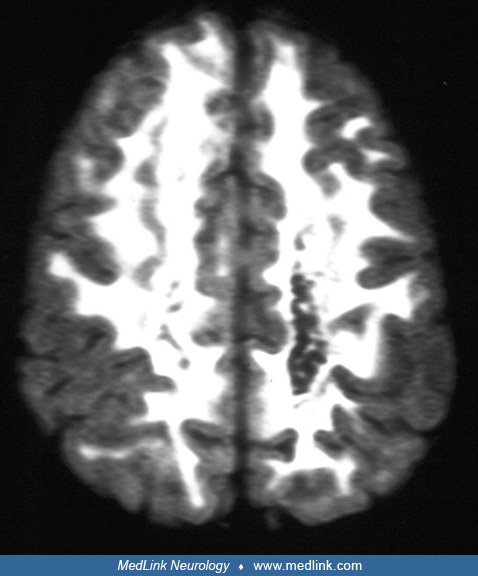
The MRI is distinctive with a diffuse decreased signal in the white matter on T1-weighted images and the corresponding increase in T2-weighted images that usually involves the subcortical white matter (52; 66).
This MRI abnormality is present even in asymptomatic children. Similar brain MRI and CT abnormalities are seen in patients with Cree leukoencephalopathy (26). Cystic breakdown of the white matter is seen on the proton density or fast-spin echo inversion recovery sequences (66; 63).
There is no gadolinium enhancement of the lesions on MRI. These abnormalities are not present in mild, relatively nonprogressive cases. Unlike many other leukodystrophies, cortical atrophy and ventricular dilation are typically absent in patients with classic childhood ataxia with central nervous system hypomyelination, even in rather advanced cases. Patients with the juvenile or the adult forms of the disease do develop ex vacuo ventriculomegaly due to the longstanding nature of the disease (Schiffmann et al 1997; 16). Early in life, there may be a component of delayed myelination in the subcortical areas that later develop the typical diffuse white matter abnormality (69). These areas are more cellular and were found to have relatively restricted diffusion on diffusion-weighted imaging studies (12; 70). Similar MRI images may be seen in mitochondrial diseases, pyruvate carboxylase deficiency, and pyruvate dehydrogenase deficiency as well as in some patients with merosin-deficient muscular dystrophy, but without extensive cavitation of the white matter as is observed in childhood ataxia with central nervous system hypomyelination (09; 29; 59; 67). Spinal cord atrophy and post-contrast enhancement with enlargement of cranial nerves has been described (Eluvathingal et al 2018). Magnetic resonance spectroscopic imaging shows a marked decrease in all metabolites over the white matter only (52; 57). However, other leukodystrophies may show a similar pattern (41). Brainstem auditory evoked potentials remain normal even in severely affected patients (57).
The association of a compatible clinical history and typical MRI with normal tests for other leukodystrophies is currently used for diagnosis (52; 63). Although characteristic pathologic findings have been seen in virtually all childhood ataxia with central nervous system hypomyelination brains examined thus far, a brain biopsy is not required to diagnose this leukodystrophy syndrome. An important development is the discovery of a significant decrease in the percent asialotransferrin of total transferrin in the CSF of EIF2B-mutated patients (73; 72). This finding may be particularly important in identifying patients with rather typical clinical and MRI abnormalities, thus, saving time and money looking for mutations affecting 1 of the 5 subunits of this protein. However, for prenatal testing, genetic testing would still be necessary.
Specific treatment is not yet available for childhood ataxia with central nervous system hypomyelination patients. Supportive therapy includes physical therapy and rehabilitation for motor dysfunction. Deep brain stimulation with electrodes implanted at the ventral intermediate nucleus of the thalamus markedly reduced cerebellar tremor in a child (60). Ankle-foot orthoses were useful in some patients with hypotonia and weakness of dorsiflexion. Carbamazepine was useful for seizures in our patients. Corticosteroids were tried in some patients, but without an impressive effect. Novel approaches based on small molecules are being developed. The most promising integrated stress response inhibitor (ISRIB), which is a molecule that stabilizes eIF2B complex and normalizes its activity (80; 01). Unfortunately, integrated stress response inhibitor is not yet available for use in humans.
Pregnancy may be a risk factor as it was in a patient when it was associated with oocyte donation (42).
Anesthesia can be performed in the usual manner in these children; however, in rare cases the neurologic status may deteriorate after general anesthesia.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Raphael Schiffmann MD
Dr. Schiffmann of Baylor Scott & White Research Institute received research grants from Amicus Therapeutics, Takeda Pharmaceutical Company, Protalix Biotherapeutics, and Sanofi Genzyme.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neurogenetic Disorders
May. 28, 2024
Neurogenetic Disorders
May. 15, 2024
Developmental Malformations
May. 15, 2024
Neurogenetic Disorders
May. 14, 2024
Neurogenetic Disorders
May. 14, 2024
Developmental Malformations
May. 14, 2024
Neurogenetic Disorders
May. 14, 2024
Neurogenetic Disorders
May. 14, 2024