






Neurobehavioral & Cognitive Disorders
Mental status examination
Jun. 17, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
The retina is the only location in the human body where morphological changes in blood vessels can be directly observed. One might say that the retina serves as a window to overall health. Fundus examination can often provide valuable information when assessing patients with neurologic and systemic diseases. It is not uncommon for patients with systemic conditions to initially present with ocular findings. In fact, systemic diseases are sometimes diagnosed based on ocular findings.
Retinal vascular disorders can manifest in many forms. The pathology primarily affects the arterial system, the venous system, or both. Some conditions, such as diabetic retinopathy, mainly involve the capillaries. These disorders are often associated with systemic conditions like hypertension, diabetes mellitus, or hypercoagulable states. Each condition has a different etiology, necessitating distinct work-up and management plans with varying levels of urgency. The primary goals in managing patients with retinal vascular disorders are preserving vision and preventing morbidity and mortality by identifying and monitoring the underlying systemic conditions.
This article aims to provide a comprehensive review of retinal vascular diseases for practicing neurologists. We will discuss the relevant vascular anatomy of the eye, common retinal vascular disorders, including hypertensive retinopathy, diabetic retinopathy, retinal artery and vein occlusions, and current management recommendations.
|
• A comprehensive understanding of retinal anatomy provides insight into the pathogenesis and clinical findings of retinal vascular diseases. | |
|
• Central retinal artery occlusion is an ocular emergency. Prompt detection and treatment are crucial for preserving vision. | |
|
• Diabetic retinopathy can potentially lead to severe vision loss due to vitreous hemorrhage, tractional retinal detachment, and neovascular glaucoma. | |
|
• Addressing systemic risk factors, such as hypertension, diabetes, and hypercholesterolemia, is crucial in managing retinal vascular disorders. |
The retina is part of the central nervous system and consists of multiple cellular layers. The outermost layer of the retina is the retinal pigment epithelium. The outer nuclear layer houses the nuclei of the photoreceptors, including rod and cone cells. The inner nuclear layer primarily comprises bipolar cells responsible for transmitting and integrating electrical signals from the photoreceptors. The innermost nuclear layer of the retina is the ganglion cell layer, from which axons extend to the optic nerve head and form nerve bundles in the optic nerve. These axons continue intracranially to synapse at the lateral geniculate nucleus.
The retina possesses a unique vascular anatomy, receiving dual circulation from both the central retinal artery system and the ciliary artery system.
The eye’s primary source of vascular supply is the ophthalmic artery, originating as the first branch of the internal carotid artery (30). After branching off from the internal carotid artery, the ophthalmic artery enters the orbit through the optic canal alongside the optic nerve. As it approaches the eye, the ophthalmic artery gives rise to the central retinal artery, which penetrates the optic nerve sheath at approximately 10 mm behind the eye, running in the center of the optic nerve. On entering the eye at the optic nerve head, the central retinal artery branches into multiple retinal arterioles, providing blood supply to all quadrants of the retina. These arterioles will eventually give rise to two layers of retinal capillaries plexus, one in the nerve fiber layer and one in the inner nuclear layer. These capillaries contain tight junctions between their endothelial cells, serving as the blood-retinal barrier. The central retinal artery system supplies the inner two thirds of the retina.
In addition to the central retinal artery system, the ophthalmic artery gives rise to 10 to 20 short ciliary arteries, primarily supplying the posterior choroid (ie, the choroid around the optic nerve head and the macula). Long ciliary arteries extend anteriorly to provide blood supply to the choroidal tissue and ciliary body. The ciliary arteries provide perfusion to the capillary bed in the innermost part of the choroid, adjacent to the retina, known as the choriocapillaris. The choriocapillaris shares its basement membrane, called Bruch’s membrane, with the retinal pigmented epithelium. The ciliary vessel system supplies the outer one third of the retina.
Pathophysiology. As part of autoregulation, an increase in systemic arterial pressure triggers vasoconstriction of the arteries to maintain a consistent perfusion level. Moreover, elevated arterial pressure exerts more force against the vessel walls, leading to physiological wall thickening, a phenomenon known as intimal hyperplasia, to withstand the increased pressure (08; 22).
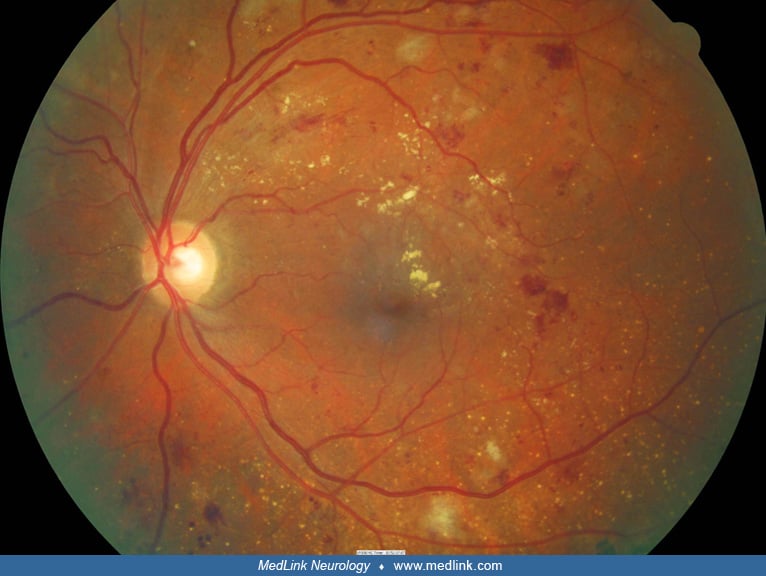
Therefore, the initial sign of hypertensive retinopathy is retinal artery vasoconstriction. Under normal circumstances, the ratio between the size of the retinal artery and the retinal vein is typically 2:3. A ratio of less than 2:3 indicates retinal artery narrowing (34). With the reduced lumen, the retinal arteries can appear to have a more yellow-orange light reflex, known as a “copper-wire” appearance. More severe luminal narrowing and wall thickening can result in a “silver-wire” appearance, signifying an extremely narrow lumen through which blood can barely pass.
Severe vasoconstriction can lead to ischemia of retinal capillaries within the nerve fiber layer, clinically manifesting as small yellow-white patches on the retinal surface, known as “cotton-wool spots” (14).
Excessively high pressure can result in the rupture of retinal capillaries in the retinal nerve fiber layer and inner nuclear layer. Hemorrhage in the nerve fiber layer gives rise to flame-shaped hemorrhages due to the parallel arrangement of nerve fibers with the retinal surface, whereas bleeding from the inner capillary plexus in the inner nuclear layer results in a dot-blot hemorrhage because the retinal cells arrange in a vertical fashion relative to the retinal surface.
In addition to bleeding, capillary ruptures also cause leakage of serum proteins and lipids into the retinal tissue, called “hard exudates.” Clinically, hard exudates appear as yellow dots or lines within the retina (35). The presence of hard exudates in the foveal region can lead to substantial vision loss.
Patients with hypertensive retinopathy typically have bilateral, symmetrical ocular involvement. Vision loss is attributable to foveal hard exudates, cystoid macular edema, macular ischemia, or optic disc ischemia.
Clinical manifestations. Patients with hypertensive retinopathy can exhibit a range of fundus findings, depending on the stage of the disease. The most widely used classification system is the Modified Scheie classification, which categorizes hypertensive retinopathy into four grades based on severity of clinical findings on dilated fundus examination:
|
Grade |
Ocular findings |
|
I |
Mild arterial narrowing |
|
II |
Obvious artery narrowing “copper-wire appearance” |
|
III |
Exudate, cotton-wool spots, retinal hemorrhage |
|
IV |
Optic nerve head swelling |
Management. The primary treatment for hypertensive retinopathy is blood pressure control (18; 13). It is crucial to emphasize that patients with hypertensive retinopathy grade III and IV, particularly those with significantly elevated blood pressure, should be viewed as having evidence of end-organ damage. In such cases, it is essential to promptly initiate a thorough work-up and management, considering the possibility of a hypertensive emergency.
Routine screening eye examinations for hypertensive retinopathy in all patients with hypertension are not generally recommended. However, for individuals with elevated blood pressure and suspected retinal involvement or visual symptoms, ophthalmological evaluation may be necessary to assess the extent of retinal damage and guide further management.
Pathophysiology. Diabetes mellitus primarily affects the capillaries. Uncontrolled serum blood sugar levels lead to nonenzymatic glycosylation of cellular proteins, ultimately giving rise to advanced glycation end-products. The formation of advanced glycation end-products results in the loss of capillary pericytes, thickening of capillary basement membranes, and increased vascular permeability, causing a breakdown of the blood-retinal barrier (31; 24).
Increased vascular permeability leads to transudation of the serum into the retina, causing retinal edema concentrated in the macular region, known as “diabetic macular edema.” The most common cause of vision loss in patients with diabetic retinopathy, diabetic macular edema develops at any stage of the disease (27).
Capillaries with weakened walls can develop microaneurysms, dot-blot hemorrhages, flame-shaped hemorrhages, and hard exudates. A decreased number of capillaries will, in turn, cause ischemia of the retinal fiber layer, sometimes manifesting as cotton-wool spots.
The more severe stage of diabetic retinopathy is called “proliferative diabetic retinopathy.” Due to the lack of capillary blood supply, the nonperfused retina responds by producing vascular endothelial growth factor to stimulate angiogenesis. The increased level of VEGF will promote the formation of fibrovascular tissue, a process known as neovascularization. Neovascularization occurs commonly at the optic disc, elsewhere in the retina, and on the surface of the iris.
Neovascularization can lead to several ocular complications, the most common being vitreous hemorrhage, due to the fragility of the new blood vessels. Patients with vitreous hemorrhage often present with sudden-onset unilateral painless vision loss and floaters. The fibrovascular membranes typically grow along the inner surface of the retina. The fibrous component of the membrane may contract, creating tractional force on the retina, which can lead to tractional retinal detachment, which is a separation of the neurosensory portion of the retina from its retinal pigmented epithelium (32).
Neovascularization of the iris and iridocorneal angle can block the aqueous outflow through the trabecular meshwork, causing an increase in the intraocular pressure, a phenomenon known as “neovascular glaucoma.”
Clinical manifestations. Diabetic retinopathy can be broadly classified into two major forms: nonproliferative diabetic retinopathy and proliferative diabetic retinopathy; the distinction is based on the presence of neovascularization (05).
|
Classification |
Ocular findings |
|
|
Nonproliferative diabetic retinopathy | ||
|
• Mild NPDR |
Microaneurysm | |
|
• Moderate NPDR |
Dot-blot hemorrhage, hard exudates, cotton-wool spots | |
|
• Severe NPDR |
One of the following: | |
|
Proliferative diabetic retinopathy |
Neovascularization of the disc, neovascularization elsewhere, neovascularization of the iris |
Management. Systemic control of blood glucose levels, blood pressure, and lipid levels is a mainstay in diabetic retinopathy management. Elevated blood glucose and hemoglobin A1C levels are associated with higher severity and rapid progression of diabetic retinopathy (12; 29).
Patients with nonproliferative diabetic retinopathy usually require periodic monitoring, the frequency of examinations varying from every 3 to 12 months, depending on the stage of nonproliferative diabetic retinopathy. Treatment for diabetic macular edema is initiated when present, as diabetic macular edema is a major cause of vision loss. Effective management of diabetic macular edema often involves intravitreal anti-VEGF injection, typically administered monthly. Focal or grid laser photocoagulation targeted to the area of capillary leakage can also be applied.
In patients with proliferative diabetic retinopathy, panretinal photocoagulation is typically performed to reduce the production of VEGF from the ischemic retina, thereby arresting the progression of neovascularization (33). Surgical management, such as pars plana vitrectomy, plays a role in cases of nonresolving vitreous hemorrhage and in eyes with tractional retinal detachment.
Pathophysiology. Central retinal artery occlusion results from emboli or thrombosis occurring at the level of lamina cribrosa. Central retinal artery occlusion is often referred to as a “stroke of the eye” and is considered an ocular emergency. Prompt management is crucial because the nonperfused retina is at a high risk of retinal infarction. Irreversible retinal damage can occur after just 90 minutes of complete retinal artery occlusion (17).
In some patients, inflammation of the artery, known as arteritis, can be the underlying cause of central retinal artery occlusion. Notably, giant cell arteritis should be considered a potential cause in elderly individuals presenting with central retinal artery occlusion.
In approximately 20% of the population, a cilioretinal artery originates from the choroidal circulation and provides blood supply to the macula. Vision can be preserved in patients with central retinal artery occlusion who have a cilioretinal artery (04).
Clinical manifestations. Patients with central retinal artery occlusion often have vascular risk factors that predispose them to the development of thrombosis and emboli formation, for example, hypertension, diabetes mellitus, hypercholesterolemia, and smoking. They typically present with sudden-onset, painless, and severe monocular vision loss. A relative afferent pupillary defect is observed.
On fundus examination, the retina shows diffuse whitening, a reflection of an edematous inner retina. However, the foveal center, which is solely supplied by the choroidal circulation, remains relatively uninvolved. The contrast between the edematous retina surrounding the relatively unaffected foveal center gives rise to the characteristic “cherry red spot,” where the pink-red color of the choroidal circulation in the foveal center is surrounded by the edematous retina.
In some cases, emboli can be visible in the central retinal artery at the optic nerve head. The appearance of the embolus can suggest its origin. For example, the detection of yellow cholesterol (Hollenhorst) or platelet-fibrin plaque typically indicates its source from cervical carotid atherosclerosis. A white calcific plaque suggests an origin from a cardiac valve.
Optical coherence tomography is a valuable diagnostic tool used to assess the cross-sectional image of the retina. In central retinal artery occlusion, the ischemic inner retina, which is supplied by retinal circulation, appears thickened and edematous, seen as hyperreflectivity of the inner retina. The outer retina remains unaffected as it is supplied by the choroidal circulation. In long-standing central retinal artery occlusion cases, OCT may reveal thinning or atrophy of the inner retina and a preserved outer retina.
Fundus fluorescein angiogram involves the injection of a dye into a vein in the patient’s arm to study the ocular circulation. The travel time and the patterns of the dye filling in the retinal arteries and veins can be studied. In central retinal artery occlusion, decreased perfusion or nonperfusion of the retinal artery and a delay in retinal artery filling time can be observed.
Management. An urgent stroke evaluation typically involves blood tests, CTA of the cervical carotid artery, electrocardiogram, echocardiogram, and brain MRI (and perhaps MRA).
For ocular management, various approaches have been studied to reduce intraocular pressure, believing that reduced intraocular pressure increases retinal perfusion (perfusion pressure = mean arterial pressure – intraocular pressure). However, no treatments are proven to significantly improve visual outcomes in central retinal artery occlusion cases. These methods include the use of acetazolamide to decrease aqueous humor production, ocular massage to increase aqueous outflow through the trabecular meshwork, anterior chamber paracentesis to remove aqueous humor from the eye, and inhalation of carbogen (95% oxygen and 5% carbon dioxide) in the hope of causing retinal vasodilation (01).
In patients with visual symptom onset within 4.5 hours and without contraindication, treatment with intravenous or intra-arterial antifibrinolytic agents has been attempted. However, results have not shown conclusive visual benefit, and costs and risks are considerable (11; 09; 28).
It is important to consider the possibility of giant cell arteritis in elderly individuals with central retinal artery occlusion. Giant cell arteritis symptoms, including fever, weight loss, polymyalgia rheumatica, jaw claudication, and scalp tenderness, should be sought. Erythrocyte sedimentation rate and C-reactive protein levels are commonly performed as screening tests to assess the possibility of giant cell arteritis.
Fluorescein angiogram can assist in diagnosing giant cell arteritis. Giant cell arteritis can involve the ophthalmic artery, which supplies the choroidal and retinal circulations. Involvement of the choroidal circulation, seen as choroidal drop-out areas in fluorescein angiogram, is suggestive of giant cell arteritis. In cases with a high suspicion of giant cell arteritis, temporal artery biopsy should be considered.
Giant cell arteritis can potentially affect the contralateral eye and lead to permanent binocular vision loss. In cases with high suspicion of giant cell arteritis, high-dose corticosteroids should be considered before confirmation of the diagnosis. It is generally accepted that temporal artery biopsy can be performed within the first 2 weeks of corticosteroid treatment without significantly altering the biopsy result (25).
Pathophysiology. Occlusion of a branch of the retinal artery is often attributed to emboli, but there are many other causes. Emboli are occasionally observable during a fundus examination, typically appearing near the point of arterial bifurcation. Common sources are the wall of the cervical carotid bifurcation, aortic arch, and heart wall or valves.
An important alternative cause of branch retinal artery occlusion is retinal vasculitis, either infectious or noninfectious inflammatory. In the elderly, giant cell arteritis should also be considered in the differential diagnosis. Multiple branch retinal artery occlusions can be a manifestation of Susac syndrome, which includes brain infarctions and cochlear deafness (15).
Clinical manifestations. Patients usually present with acute onset scotoma in the superior or inferior visual field. The symptom can sometimes be unnoticeable to the patient, especially when the infarction does not involve the fovea. Clinically, sectoral retinal edema, seen as whitening of the retina, and sectoral narrowing of the retinal artery can be observed (10).
A fluorescein angiogram can help in diagnosing branch retinal artery occlusion. Sectoral decrease in arterial perfusion, delayed arterial filling time, and retinal artery narrowing can be appreciated. OCT through the involved area demonstrates thickening and hyperreflectivity of the inner retina with normal outer retina.
Management. The source of emboli should be sought promptly to prevent further embolism to other organs (20; 16). Nonembolic vasculopathies must be ruled out, especially in younger individuals.
Pathophysiology. Thrombosis of the central retinal vein at the level of the lamina cribrosa is the main mechanism of central retinal vein occlusion. It usually occurs in older patients with essential hypertension, hyperlipidemia, or diabetes mellitus. Compression of the central retinal vein by an arteriosclerotic central retinal artery results in turbulent flow inside the venous lumen, endothelial injury, and thrombosis (26).
Central retinal vein occlusion is sometimes associated with hypercoagulable states, such as antiphospholipid syndrome, multiple myeloma, Waldenstrom macroglobulinemia, and other blood dyscrasias. Investigations for these entities should be considered in young populations and in patients with bilateral central retinal vein occlusion.
Clinical presentation. Patients typically present with acute to subacute onset unilateral painless vision loss (21). Fundus examination shows dilated and tortuous retinal veins caused by the back pressure of blood that cannot drain through the central retinal vein. The clinical hallmark of central retinal vein occlusion is the nerve fiber layer (flame-shaped) hemorrhage, but the most common causes of visual loss are cystoid macular edema, macular ischemia, and foveal hard exudates.
A long-term consequence of central retinal vein occlusion is neovascular glaucoma, which typically occurs within 3 months after the onset (“90-day-glaucoma”). In response to ischemia, the retina produces VEGF, which subsequently promotes the growth of neovascular membranes on the retina, iris, and iridocorneal angle, leading to aqueous outflow blockage and, eventually, neovascular glaucoma.
Management. The visual prognosis in patients with central retinal vein occlusion correlates with the degree of retinal ischemia. Fluorescein angiography can effectively demonstrate the area of retinal ischemia. Optical coherence tomography of the macula can be used to evaluate and monitor cystoid macular edema.
The treatment of central retinal vein occlusion is directed at reducing cystoid macular edema and monitoring for neovascularization. Intravitreal anti-VEGF has been proven effective in reducing macular edema and improving vision (07; 02; 19). Laser photocoagulation is effective in preventing and reversing neovascularization. By destroying portions of the peripheral retina, it decreases the production of VEGF.
Systemic management focuses on controlling vascular risk factors and underlying diseases.
Pathophysiology. In branch retinal vein occlusion, venous obstruction usually occurs at the arteriovenous crossing, most commonly in the superotemporal quadrant. In normal circumstances, the retinal artery and vein share an adventitia at the crossing. Thickening and hardening of the arterial wall in arteriosclerosis can compress the collapsible wall of the vein (23). The pathophysiology of branch retinal vein occlusion is strongly associated with hypertension. Less common causes of branch retinal vein occlusion include infectious and noninfectious inflammatory etiologies.
Clinical manifestations. Patients with branch retinal vein occlusion typically present with unilateral painless vision loss, often described as seeing a scotoma in the superior or inferior visual field. Some patients are asymptomatic. Fundus examination shows dilated and tortuous veins with nerve fiber layer hemorrhages in one quadrant of the retina. Vision loss can also be due to cystoid macular edema, macular exudates, or macular ischemia. Neovascularization of the retina can develop secondary to ischemia, but neovascularization of the iris and iridocorneal angle are uncommon.
Management. Cystoid macular edema is usually treated effectively with intravitreal injection of anti-VEGF agents (06; 03). Macular grid laser photocoagulation is an alternative treatment. In cases with retinal neovascularization, sectoral laser retinal photocoagulation can be considered.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Chaow Charoenkijkajorn MD
Dr. Charoenkijkajorn of Methodist Eye Associates in Houston, Texas, has no relevant financial relationships to disclose.
See Profile
Jonathan D Trobe MD
Dr. Trobe of the University of Michigan has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neurobehavioral & Cognitive Disorders
Jun. 17, 2026
Neuro-Oncology
May. 27, 2026
Neuro-Oncology
May. 27, 2026
Neuropharmacology & Neurotherapeutics
May. 14, 2026
General Child Neurology
May. 12, 2026
Neuro-Oncology
Apr. 30, 2026
Neuropharmacology & Neurotherapeutics
Apr. 23, 2026
Neuropharmacology & Neurotherapeutics
Apr. 20, 2026