





Methylmalonic acidemia
Jun. 01, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
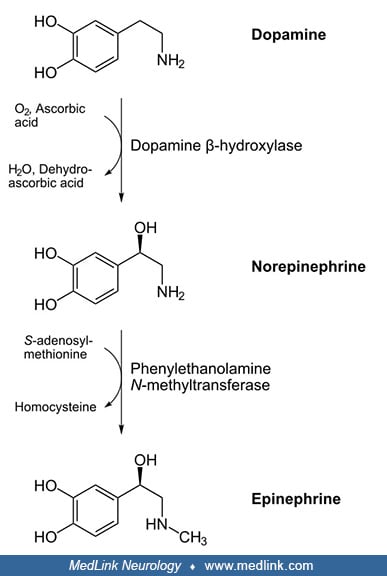
Dopamine beta-hydroxylase deficiency is an autosomal recessive genetic form of primary autonomic failure. Dopamine beta-hydroxylase normally converts dopamine to norepinephrine (which can then be further metabolized to epinephrine), so a deficiency or dysfunction of this enzyme causes a profound deficiency of norepinephrine and epinephrine within the central and peripheral nervous systems. Patients with dopamine beta-hydroxylase deficiency have generally been detected in adolescence or adulthood following the onset of severe orthostatic hypotension. Although symptoms are usually diagnosed in adulthood, ptosis, hypothermia, severe orthostatic hypotension, hypoglycemia, and other autonomic abnormalities may have been evident since birth. Testing reveals extremely low levels of norepinephrine, epinephrine, and their metabolites in plasma, CSF, and urine. Treatment with the norepinephrine prodrug droxidopa (dl-dihydroxyphenylserine, L-DOPS) increases plasma and urine norepinephrine to near normal levels, with a consequent increase in blood pressure and amelioration or correction of the symptoms of orthostatic hypotension.
|
• Dopamine beta-hydroxylase deficiency is inherited as an autosomal recessive trait. | |
|
• Dopamine beta-hydroxylase deficiency leads to a profound deficiency of norepinephrine and epinephrine within the central and peripheral nervous systems. | |
|
• Dopamine beta-hydroxylase deficiency leads to a primary autonomic failure. | |
|
• Dopamine beta-hydroxylase deficiency has only a minor impact on cognitive function. | |
|
• Treatment with the norepinephrine prodrug droxidopa (dl-dihydroxyphenylserine, L-DOPS) increases plasma and urine norepinephrine to near normal levels, with a consequent increase in blood pressure and amelioration or correction of the symptoms of orthostatic hypotension. |
Dopamine beta-hydroxylase was discovered in the 1950s (20). Dopamine beta-hydroxylase is a copper-containing, ascorbate-dependent enzyme that synthesizes norepinephrine from dopamine (20). Subsequent methylation of norepinephrine (nonadrenaline) forms epinephrine (adrenaline), and these two catecholamine neurotransmitters are the main determinants of vascular tone and, hence, arterial pressure.
In 1986 and 1987, two groups reported a syndrome of autonomic failure characterized by severe orthostatic hypotension, noradrenergic failure, and ptosis of the eyelids but with normal parasympathetic function and normal sympathetic cholinergic function (47; 34). The biochemical findings of low or absent norepinephrine and epinephrine and their metabolites in the presence of elevated dopamine concentrations, combined with the clinical data, led to the conclusion that this condition was due to a congenital deficiency of dopamine beta-hydroxylase. Activity of this enzyme was absent in plasma. Earlier cases with a similar syndrome have also been reported, but a definitive diagnosis was not made (02; 40).
Patients with dopamine beta-hydroxylase deficiency have generally been detected in adolescence or adulthood following the onset of severe orthostatic hypotension. The mode of inheritance is autosomal recessive, and several different mutations have been identified (18; 11).
|
• Dopamine beta-hydroxylase deficiency is characteristically diagnosed in late childhood or adulthood with the appearance of severe orthostatic hypotension and noradrenergic failure. | |
|
• Retrospective case histories have demonstrated difficulties in the perinatal period, including delay in opening of the eyes, ptosis of the eyelids, hypotension, hypothermia, and hypoglycemia. | |
|
• Symptoms of the disease progressively worsen with age. | |
|
• Due to profound orthostatic hypotension, patients usually cannot stand motionless for more than 30 seconds. | |
|
• Other manifestations can include mild eyelid ptosis, nasal stuffiness, reduced exercise tolerance, and prolonged or retrograde ejaculation. |
Almost all cases of dopamine beta-hydroxylase deficiency have been diagnosed in late childhood or adulthood with the appearance of severe orthostatic hypotension and noradrenergic failure (60; 04). Dopamine beta-hydroxylase deficiency has rarely been recognized in early life (36), and the spectrum of clinical features in the neonatal period and in infancy is poorly understood. A premature female newborn who was delivered via cesarean section at 26 weeks of gestation owing to profound bradycardia and intrauterine growth restriction during pregnancy developed repetitive syncope in the early postnatal period leading to pacemaker implantation and ultimately a diagnosis of dopamine beta-hydroxylase deficiency (36). Retrospective case histories of cases identified at older ages have demonstrated difficulties in the perinatal period, including vomiting, dehydration, delay in opening of eyes, ptosis of the eyelids, hypotension, hypothermia, and hypoglycemia (48; 17; 39). It is still unclear whether the patients surviving into adulthood are typical or whether they represent the "best case scenario," with more severe forms of the disease being incompatible with life. Long-term survival, even without treatment, is possible, as demonstrated by a 73-year-old man with the disorder who has had untreated orthostatic hypotension since childhood (12).
A history of spontaneous abortions and stillbirths is reported in mothers of affected patients, but available reports are so limited that controlled studies have not been done to prove these occur at abnormal frequencies (34; 48). However, mortality in homozygous embryos from "knockout" dopamine beta-hydroxylase-deficient mice is high, with only 5% of progeny reaching adulthood (56). Norepinephrine-deficient mice also are highly susceptible to seizure-inducing stimuli and have altered cellular immunity (01; 55).
Symptoms of the disease progressively worsen with age. Children have had postural hypotension and syncope (47; 37). In adolescence and early adulthood, there are striking orthostatic reductions in intraocular pressure and mean arterial blood pressure (43). Blood pressure is low to normal in the supine position (48), but due to profound orthostatic hypotension (falling below 80 mm Hg on standing with a compensatory increase in heart rate), patients usually cannot stand motionless for more than 30 seconds. In many cases, there is also mild eyelid ptosis, nasal stuffiness, reduced exercise tolerance, and prolonged or retrograde ejaculation (05; 37; 48; 43). By early adulthood, individuals have profound orthostatic hypotension, greatly reduced exercise tolerance, eyelid ptosis, and nasal stuffiness (17; 04). Presyncopal symptoms are typical but not specific to this disorder: they include dizziness, blurred vision, dyspnea, nuchal discomfort, and chest pain; symptoms may worsen in hot environments or after heavy meals, exercise, or alcohol ingestion (17).
The various autonomic manifestations are indicative of sympathetic adrenergic failure (17):
|
• Severely symptomatic orthostatic hypotension, with a compensatory rise in heart rate with standing. | |
|
• Somewhat small pupils that respond to light and accommodation but not to hydroxyamphetamine. | |
|
• Parasympatholytics dilate the pupils appropriately. | |
|
• Eyelid ptosis | |
|
• Nasal stuffiness | |
|
• Intact sweating consistent with intact sympathetic cholinergic function. | |
|
• Retrograde ejaculation in males. | |
|
• The Valsalva maneuver results in a profound fall in blood pressure and an increase in heart rate, reflecting parasympathetic withdrawal. |
Other variable clinical manifestations include high palate, hyperextensible or hyperflexible joints, brachydactyly, nocturia, mild behavioral changes, seizures following hypotension, hypothermia, vomiting, dehydration, weak facial musculature, hypotonic skeletal muscles, sluggish deep tendon reflexes, impairment in cardiovascular autonomic regulation, atrial fibrillation, and T-wave abnormalities on EKG (48; 04). Variable metabolic abnormalities include hypomagnesemia, raised blood urea nitrogen, hypoprolactinemia, hyperinsulinemia, enhanced glucose-stimulated insulin secretion, and insulin resistance; these metabolic derangements were not corrected by chronic droxidopa treatment (48; 03; 04).
Sleep duration is normal, but there is a decreased amount of rapid eye movement sleep, which is corrected after administration of dihydroxyphenylserine (58).
Individuals with dopamine beta-hydroxylase deficiency do not have major cognitive deficits. A systematic investigation of neurocognitive function in five patients with dopamine beta-hydroxylase deficiency, performed both on and off of medication, found no substantial deficit in cognitive performance except a temporal-attention deficit in individuals off of medication (22).
Life expectancy is unknown, but some affected individuals have lived beyond 60 years of age (12; 17).
A single woman had no symptoms until the age of 61 years. Idiopathic dysautonomic orthostatic hypotension was diagnosed at this time (19). Her condition worsened, and falls became frequent, resulting in three femoral fractures over a period of 3 years. A transvenous coronary-sinus atrial demand pacemaker was implanted with temporary improvement; however, the patient became bedridden at the age of 79 years. At age 83, supine blood pressure was 120/60 mmHg. On standing, systolic pressure fell to 40 mmHg, and diastolic pressure could not be measured. Plasma catecholamine analysis was consistent with dopamine beta-hydroxylase deficiency, and confirmation of the diagnosis was obtained by discovery of low dopamine beta-hydroxylase activity in plasma. Treatment with droxidopa (dl-threo-dihydroxyphenylserine, L-DOPS) normalized plasma norepinephrine levels, increased upright blood pressure to 90/60mmHg, and allowed her to walk with assistance.
|
• Dopamine beta-hydroxylase deficiency is an autosomal recessive disorder due to a mutation in the DBH gene on chromosome 9q34. | |
|
• Dopamine beta-hydroxylase catalyzes the conversion of dopamine to norepinephrine (by hydroxylation of dopamine); norepinephrine can then be methylated via the action of phenylethanolamine N-methyltransferase to form epinephrine. | |
|
• Deficiency of dopamine beta-hydroxylase leads to the virtual absence of norepinephrine and epinephrine and their metabolites (ie, vanillylmandelic acid, 3-methoxy-4-hydroxyphenylglycol, and normetanephrine) and to elevations of L-dopa, dopamine, and metabolites (homovanillic acid, dihydroxyphenylacetic acid) in plasma, urine, and CSF. | |
|
• Orthostatic hypotension, the major feature of the disease, is caused by the lack of a pressor effect from norepinephrine and epinephrine, which in the periphery are the main determinants of vascular tone and, hence, arterial pressure. |
Dopamine beta-hydroxylase deficiency is an autosomal recessive disorder (18) due to a mutation in the DBH gene on chromosome 9q34 (26).
Various mutations in the dopamine beta-hydroxylase gene have been described that are associated with alterations in dopamine beta-hydroxylase activity (62; 18; 30; 11; 29). Mutations associated with disease generally produce truncated proteins or an altered sequence in highly conserved areas (11). An IVS1+2T>C splice site mutation results in nondetectable dopamine beta-hydroxylase protein, and various missense mutations cause the defective dopamine beta-hydroxylase protein to be trapped in the endoplasmic reticulum (29). A homozygous G>T conversion in exon 4 leads to substitution of cysteine by phenylalanine in a highly preserved region for copper-containing, ascorbate-dependent mono-oxygenases (11).
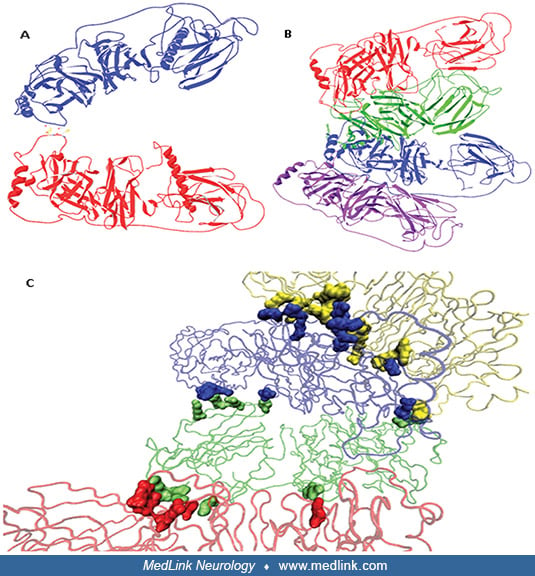
Dopamine beta-hydroxylase is a 290 kDa copper-containing oxygenase composed of four identical subunits. Dopamine beta-hydroxylase is expressed in noradrenergic nerve terminals of the central and peripheral nervous systems as well as in chromaffin cells of the adrenal medulla.
Dopamine beta-hydroxylase catalyzes the conversion of dopamine to norepinephrine (by hydroxylation of dopamine). Norepinephrine can then be methylated via the action of phenylethanolamine N-methyltransferase to form epinephrine.
Because dopamine beta-hydroxylase is the only membrane-bound enzyme involved in synthesizing of small-molecule neurotransmitters, norepinephrine is the only transmitter synthesized inside vesicles. The conversion of dopamine to norepinephrine by dopamine beta-hydroxylase occurs via hydroxylation of the beta carbon of the ethylamine moiety of dopamine.
This conversion depends on the use of ascorbate (vitamin C) as a cofactor.
Dopamine beta-hydroxylase also catalyzes the hydroxylation of other phenylethylamine derivatives (ie, a benzene ring with a 2-carbon side chain that terminates in an amino group) when available. Dopamine beta-hydroxylase requires two coppers per subunit for optimal activity (31). Both coppers are involved in the catalytic mechanism separate binding sites for reductants and product/substrate and, hence, separate functions for each copper per subunit (31). The reaction apparently proceeds in two steps (only the first of which involves ascorbate): first, the enzyme and ascorbate react to produce reduced enzyme and dehydroascorbate; then, the reduced enzyme reacts with oxygen and dopamine, which results in oxidation of the enzyme and formation of norepinephrine (noradrenaline) and water.
Deficiency of dopamine beta-hydroxylase leads to the virtual absence of norepinephrine and epinephrine and their metabolites (ie, vanillylmandelic acid [VMA], 3-methoxy-4-hydroxyphenylglycol [MHPG], and normetanephrine [NMN]) and to elevations of L-dopa, dopamine, and dopamine metabolites (eg, dihydroxyphenylacetic acid [DOPAC] and homovanillic acid [HVA]) in plasma, urine, and CSF (47; 35). Dopamine and L-dopa are elevated for two reasons: (1) dopamine is stored in noradrenergic neurons instead of norepinephrine and is released as if it were norepinephrine; and (2) norepinephrine normally acts as a feedback inhibitor of tyrosine hydroxylase, the rate-limiting enzyme for dopamine biosynthesis, but in the absence of norepinephrine, tyrosine hydroxylase remains active, leading to higher steady-state concentrations of dopamine.
Orthostatic hypotension, the major feature of the disease, is caused by the lack of a pressor effect from norepinephrine and epinephrine, which in the periphery are the main determinants of vascular tone and, hence, arterial pressure. Instead of norepinephrine, dopamine is stored in the sympathetic noradrenergic terminals, and because central autonomic control, dopamine synthesis, and catecholamine-release mechanisms remain intact, appropriate stimuli lead to release of the “wrong” neurotransmitter (ie, dopamine release instead of norepinephrine). The increase in dopamine contributes to the drop in blood pressure, as dopamine has a vasodepressor effect that occurs either through direct vasodilation or by means of a diuretic effect in the kidney (13; 32). The circadian rhythm in blood pressure is also reversed in dopamine beta-hydroxylase deficiency and a higher blood pressure at night contributes to pressure natriuresis (58). This, together with the diuretic effect from the high dopamine concentration, probably explains the nocturia these patients experience.
The ptosis of the eyelids reflects failed noradrenergic control of levator function (47).
Hypoglycemia and hypothermia during infancy have not been fully explained. Robertson suggested that hypoglycemia results from the lack of a calorigenic effect in the absence of epinephrine and that the elevated dopamine levels contribute to hypothermia. Evidence from animal experiments has demonstrated that excessive dopamine causes temperature reduction (48).
The pathologic changes in dopamine beta-hydroxylase deficiency are due to chronic autonomic failure. Magnetic resonance imaging scans of the brain have revealed a decrease in brain volume, suggesting norepinephrine may have a neurotrophic effect (22). Autopsy of a 28-year-old man with dopamine beta-hydroxylase deficiency did not reveal any major structural abnormalities in the brain, but immunostaining showed a total lack of dopamine beta-hydroxylase enzyme in the ventrolateral medulla, demonstrating involvement of the central autonomic network (22). Studies in brain, CSF, plasma, and tissue have shown a lack of immunoreactive protein (37; 41; 08). The sympathetic nerves themselves remain intact (45; 12), although sympathetic-nerve firing rates in muscle are increased as shown by microneurography (57).
A population-based analysis found an association between the 19-bp insertion/deletion polymorphism upstream of the DBH gene and autism spectrum disorder but only when the analysis was performed on male participants (61); no association at the genotype level was found.
DBH polymorphisms have been associated with susceptibility to some neurologic diseases, such as Alzheimer disease, but the evidence to date is weak (24).
Dopamine beta-hydroxylase deficiency is a very rare disorder. No epidemiological information is available.
|
• Dopamine beta-hydroxylase deficiency is inherited as an autosomal recessive disorder. | |
|
• Parents of an affected individual are usually asymptomatic carriers. | |
|
• Early recognition is important to prevent significant morbidity and mortality because the disease can now be treated successfully by administration of the prodrug droxidopa. |
Dopamine beta-hydroxylase deficiency is inherited as an autosomal recessive disorder. Thus, parents of an affected individual are usually asymptomatic carriers, each with one abnormal gene: each pregnancy for such a couple has a 1:4 chance of producing an affected child. Once the DBH pathogenic variants have been identified in an affected family member, carrier testing should be performed for at-risk relatives (eg, siblings) and prenatal or preimplantation genetic testing are also possible (04).
Early recognition is important to prevent significant morbidity and mortality because the disease can now be treated successfully by administration of the prodrug droxidopa (dl-threo-3,4-dihydroxyphenylserine, L-DOPS) (06; 35).
Untreated individuals should avoid hot environments, strenuous exercise, standing motionless, and dehydration (04). Nephrotoxic drugs should also be avoided (04).
The syncope associated with postural hypotension often suggests seizures to inexperienced clinicians, particularly when there are associated brief limb jerks ("convulsive syncope") (17). Too often, this prompts trials of anticonvulsive medications, despite the apparent (though often clinically unassessed) orthostatic hypotension and the lack of abnormalities on the electroencephalogram.
The major clinical feature of dopamine beta-hydroxylase deficiency is chronic hypotension, with devastating orthostatic hypotension being characteristic in adults (50). However, a multitude of conditions can lead to acute or chronic orthostatic hypotension, including drug toxicities, autonomic neuropathies, baroreceptor dysfunction, paroxysmal autonomic syncope, endocrine disorders, vascular insufficiency, hypovolemic disorders, and neurodegenerative disorders (eg, Lewy body dementia, multisystem atrophy) (33; 50). In addition, orthostatic intolerance has been reported in a patient with a norepinephrine-transporter deficiency (53). The clinical history and physical examination will often point to the underlying cause (51; 50). It is important to exclude drug-induced orthostatic hypotension, as can occur, for example, with the use of marijuana, antihypertensive agents, diuretics, nitrates, alpha1-blockers (prazosin, doxazosin, terazosin, and tamsulosin), tricyclic antidepressants, antipsychotics, and carbidopa-levodopa and dopamine agonists (50).
Dopamine beta-hydroxylase deficiency is a primary autonomic neuropathy—a disorder in which autonomic neuropathy is a characteristic feature of the disease process itself—affecting exclusively sympathetic noradrenergic neurons. It must be distinguished from other conditions that lead to chronic autonomic nervous system failure. Other primary autonomic neuropathies include pure autonomic failure (PAF; other terms include: Bradbury-Eggleston syndrome, progressive autonomic failure; idiopathic orthostatic hypotension); a form of peripheral autonomic failure (07); multiple system atrophy (MSA; Shy-Drager syndrome), causing central autonomic failure (54); and familial dysautonomia (Riley-Day syndrome; type III hereditary sensory autonomic neuropathy; type III HSAN), affecting the development and survival of sensory, sympathetic, and parasympathetic neurons (46). In addition, secondary forms of autonomic neuropathy may develop as a feature of some systemic diseases, such as diabetes (16), acute intermittent porphyria and variegate porphyria (42), Fabry disease, and amyloidosis (50).
The Riley-Day syndrome is a familial dysautonomia almost exclusively affecting Ashkenazi Jews (46), and orthostatic hypotension is only one of the widespread abnormalities within the central and peripheral nervous systems in this disorder. These patients have poor or absent axon flare reaction to intradermal histamine, miosis after conjunctival instillation of methacholine, absence of fungiform papillae on the tongue, abnormal tear production, problems with taste and smell, decreased or absent reflexes, and poor motor coordination (38). In contrast, patients with dopamine beta-hydroxylase deficiency have a normal histamine response, no reaction to methacholine, normal tongue papillae, normal tearing, normal senses of taste and smell, intact deep tendon reflexes, and normal coordination. Furthermore, dopamine beta-hydroxylase deficiency has never been identified in Ashkenazi Jews (48).
Two families have been identified with a disorder closely resembling dopamine beta-hydroxylase deficiency (59). As in dopamine beta-hydroxylase deficiency, norepinephrine and epinephrine concentrations were low, but plasma dopamine beta-hydroxylase activity was normal, and the DBH gene had no mutations. Pathogenic homozygous mutations in the gene encoding cytochrome b561 (CYB561) were identified in both families; a missense variant c.262G>A, p.Gly88Arg in exon 3 was identified in the Dutch family, and a nonsense mutation (c.131G> A, p.Trp44*) in exon 2 was identified in the American family. The defective CYB561 protein results in a shortage of ascorbate inside the catecholamine secretory vesicles, resulting in a functional dopamine beta-hydroxylase deficiency. Like dopamine beta-hydroxylase deficiency, this disorder responds to droxidopa.
Menkes disease is a neurodegenerative disorder of copper metabolism. Because dopamine beta-hydroxylase requires copper for activity, the plasma and cerebrospinal fluid catechol pattern in Menkes disease may resemble that seen in dopamine beta-hydroxylase deficiency (25). In particular, there are high concentrations of dopamine, dopamine precursors (eg, DOPA), and dopamine metabolites (eg, DOPAC) in plasma and CSF, approximately normal concentrations of norepinephrine, and low concentrations of norepinephrine metabolites (eg, DHPG). Clinically, the two diseases are easily separated because Menkes patients usually die between 6 months and 3 years of age, have focal cerebral and cerebellar degeneration, show early growth retardation and mental retardation, and have characteristic "kinky" pale hair.
Differentiation between the primary autonomic neuropathies can be made in several ways:
|
• Inheritance: Dopamine beta-hydroxylase deficiency and Riley-Day syndrome are autosomal recessive disorders, although Riley-Day syndrome occurs almost exclusively in people of Ashkenazi Jewish descent, whereas dopamine beta-hydroxylase deficiency has never been reported in individuals of Ashkenazi Jewish descent. Genetic variants of the dopamine beta-hydroxylase gene are not responsible for either pure autonomic failure or multiple system atrophy (09). | |
|
• Biochemistry and metabolite concentrations: Biochemically, dopamine beta-hydroxylase deficiency is different from all other recognized conditions with orthostatic hypotension or autonomic dysfunction. Deficiency of dopamine beta-hydroxylase leads to the virtual absence of norepinephrine and epinephrine and their metabolites (ie, vanillylmandelic acid [VMA], 3-methoxy-4-hydroxyphenylglycol [MHPG], and normetanephrine [NMN]) and to elevations of L-dopa, dopamine, and dopamine metabolites (eg, dihydroxyphenylacetic acid [DOPAC] and homovanillic acid [HVA]) in plasma, urine, and CSF (47; 35). The combination of orthostatic hypotension, minimal or undetectable plasma norepinephrine, together with a 5- to 10-fold elevation of plasma dopamine is unique to this disease (due to the metabolic block in the conversion of dopamine to norepinephrine). In norepinephrine transporter deficiency, plasma norepinephrine levels are elevated (53). In PAF, plasma norepinephrine levels are also greatly reduced, but this occurs in combination with a reduction in dopamine (63). Levels of plasma norepinephrine may be near normal in multiple system atrophy (Shy-Drager syndrome), although CSF levels of norepinephrine and dopamine metabolites are decreased, reflecting the central nature of the disease (44). | |
|
• Age of onset: Patients with dopamine beta-hydroxylase deficiency typically present earlier in life than those with multiple system atrophy or pure autonomic failure. Symptoms of dopamine beta-hydroxylase deficiency typically appear early life and the spectrum of clinical in life, although one reported patient had symptom onset at age 61 years (19). | |
|
• Type of autonomic failure: Dopamine beta-hydroxylase deficiency and pure autonomic failure are both forms of postganglionic (efferent) autonomic failure, whereas multiple system atrophy has predominantly central involvement. | |
|
• Clinical features in the neonatal period and in infancy related to nonautonomic dysfunction: Dopamine beta-hydroxylase deficiency only affects the sympathetic noradrenergic neurons, and there is no evidence of other neurologic abnormalities. Pure autonomic failure is a degenerative disorder involving both the parasympathetic and sympathetic nervous systems (although from a clinical standpoint, the sympathetic failure is predominant). In multiple system atrophy, there are other features of cerebellar, extrapyramidal, neuromuscular, or cortical dysfunction. |
|
• In a proband with profound neurogenic orthostatic hypotension, the diagnosis of dopamine beta-hydroxylase deficiency is established by documenting minimal or absent plasma concentrations of norepinephrine and epinephrine and a five- to tenfold elevation of plasma dopamine. | |
|
• The diagnosis of dopamine beta-hydroxylase deficiency is confirmed with molecular genetic testing by the demonstration of biallelic pathogenic variants in DBH. | |
|
A beneficial response to the norepinephrine prodrug dl-dihydroxyphenylserine (L-DOPS, droxidopa) is also diagnostic. |
The diagnosis of dopamine beta-hydroxylase deficiency is often delayed (39).
In a proband with profound neurogenic orthostatic hypotension, the diagnosis of dopamine beta-hydroxylase deficiency is established by documenting minimal or absent plasma concentrations of norepinephrine and epinephrine, and a five- to ten-fold elevation of plasma dopamine; it is confirmed with molecular genetic testing by the demonstration of biallelic pathogenic variants in DBH (17; 04).
Plasma catecholamines. Measurement of plasma dopamine beta-hydroxylase activity cannot be used to make a definitive diagnosis of the disease because there is a genetically determined inter-individual variation in plasma dopamine beta-hydroxylase, with 3% to 4% of the normal adult population having very low plasma activity (15); this may be related to the presence of a polymorphism in the 5’ flanking region (C> T; -1021) of the dopamine beta-hydroxylase gene (62; 11).
In dopamine beta-hydroxylase deficiency, levels of norepinephrine and epinephrine and their metabolites are very low or absent in plasma, cerebrospinal fluid, and urine (60). In contrast, plasma dopamine concentrations are elevated 5- to 10-fold, and there is a 2- to 3-fold increase in L-dopa (60). Normally the plasma of the norepinephrine-to-dopamine ratio is around 10, but in patients with dopamine beta-hydroxylase deficiency, it is below 0.1 (48). A norepinephrine-to-dopamine ratio of less than 0.1 is probably diagnostic for the disease without further tests; however, supporting information can be obtained by measurement of catechols in urine or CSF. Reduced or absent levels of norepinephrine and its metabolites (normetanephrine, vanillylmandelic acid, and 3-methoxy-4-hydroxyphenolglycol) combined with elevated levels of L-dopa, dopamine, and its metabolites (homovanillic acid and dihydroxyphenylacetic acid) provide further supporting diagnostic evidence.
Physiologic tests of autonomic function. Physiologic tests of autonomic function demonstrate specific abnormalities of autonomic function, including (1) isometric handgrip, cold pressor testing, and mental arithmetic fail to elicit a pressor response (50); (2) tyramine fails to raise plasma norepinephrine levels and leads to a decrease, rather than an elevation, of blood pressure as occurs in other types of autonomic failure (49); (3) the Valsalva maneuver results in a fall in blood pressure and an increase in heart rate, reflecting parasympathetic withdrawal, and there is no pressure overshoot in phase IV of the maneuver, indicating disruption of the integrity of the baroreflex arc (47; 35); and (4) alpha- and beta-adrenergic antagonists fail to elicit a hemodynamic response, confirming sympathetic failure (35).
A normal sweating response, indicating the integrity of sympathetic cholinergic innervation of eccrine sweat glands and preserved respiratory sinus arrhythmia (RSA; the natural variation in heart rate that occurs during the breathing cycle), reflecting intact parasympathetic function, help distinguish dopamine beta-hydroxylase deficiency from other forms of autonomic failure where there is both sympathetic and parasympathetic involvement. Intact parasympathetic innervation and deficient sympathetic innervation can also be demonstrated in the eye, where pupil size does not change following conjunctival instillation of methacholine, homatropine, or hydroxyamphetamine (23; 34).
Skin biopsy and microneurography may be helpful in diagnosis, particularly when routine autonomic tests produce equivocal results (14).
Dopamine beta-hydroxylase deficiency has not been diagnosed in infancy but should be considered in neonates and infants with unexplained hypoglycemia, hypothermia, hypotension, or eyelid ptosis. A patient with Menkes disease might have a similar pattern of catecholamines (dopamine and norepinephrine) and their metabolites, and this disease should be excluded (25).
Molecular genetic testing. Molecular genetic testing can include a combination of gene-targeted testing (single-gene testing, multigene panel) and comprehensive genomic testing (exome sequencing, exome array, genome sequencing) depending on the phenotype (17). When the phenotypic and laboratory findings suggest the diagnosis of dopamine beta-hydroxylase deficiency, molecular genetic testing can utilize single-gene testing or a multigene panel. Sequence analysis of DBH detects small intragenic deletions/insertions and missense, nonsense, and splice site variants. Exon or whole-gene deletions/duplications are not detected. If only one or no pathogenic variant is found on sequence analysis, gene-targeted deletion/duplication analysis can detect intragenic deletions or duplications. A multigene panel that includes DBH and other genes of interest is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype.
Therapeutic trial. A beneficial response to the norepinephrine prodrug dl-dihydroxyphenylserine (L-DOPS, droxidopa) is also diagnostic (see Management).
Other laboratory tests. Other abnormalities of laboratory studies can include impaired renal function, anemia, and hypomagnesemia (60). Measurement of blood urea nitrogen and plasma creatinine to assess kidney function should be done every 2 years, or more often if loss of kidney function is evident (04); magnesium and potassium should also be checked every 2 years (04).
|
• The management of dopamine beta-hydroxylase deficiency requires correction of the norepinephrine deficit, which is achieved by administration of droxidopa (dl-dihydroxyphenylserine, L-DOPS). | |
|
• The norepinephrine prodrug droxidopa is an orally active synthetic amino acid (ie, a carboxylated derivative of norepinephrine) that is converted to norepinephrine in vivo by the enzyme aromatic L-amino acid decarboxylase (dopa-decarboxylase). | |
|
• The conversion of L-DOPS (dl-dihydroxyphenylserine, droxidopa) to norepinephrine by aromatic L-amino acid decarboxylase (dopa-decarboxylase) is analogous to the conversion of the prodrug L-DOPA (3,4-dihydroxy-L-phenylalanine, levodopa) to dopamine by the same enzyme. | |
|
• In dopamine beta-hydroxylase deficiency, droxidopa increases plasma and urine norepinephrine to near normal levels, with a consequent increase in blood pressure and amelioration or correction of the symptoms of orthostatic hypotension. |
The management of dopamine beta-hydroxylase deficiency requires correction of the norepinephrine deficit. This is achieved by administration of droxidopa (dl-dihydroxyphenylserine, L-DOPS), which alleviates the orthostatic hypotension and other symptoms of abnormal cardiovascular regulation (04).
The norepinephrine prodrug droxidopa is an orally active synthetic amino acid (ie, a carboxylated derivative of norepinephrine) that is converted to norepinephrine in vivo by the enzyme aromatic L-amino acid decarboxylase (dopa-decarboxylase) (06; 35; 27; 28).
The conversion of L-DOPS (dl-dihydroxyphenylserine, droxidopa) to norepinephrine by aromatic L-amino acid decarboxylase (dopa-decarboxylase) is analogous to the conversion of the prodrug L-DOPA (3,4-dihydroxy-L-phenylalanine, levodopa) to dopamine by the same enzyme.
In dopamine beta-hydroxylase deficiency, droxidopa increases plasma and urine norepinephrine to near normal levels, with a consequent increase in blood pressure and amelioration or correction of the symptoms of orthostatic hypotension (06; 35; 37; 19; 27; 28; 60). Orthostatic hypotension continues to be present in some patients, despite a marked reduction of orthostatic complaints (60). In addition, kidney function, anemia, and hypomagnesaemia only partially improve (60). Droxidopa is generally well tolerated with sustained benefit, but patients need to be monitored for development of supine hypertension (27; 28; 21).
Individuals with dopamine beta-hydroxylase deficiency do not respond as well to standard therapeutic approaches for autonomic failure (17).
Other treatment options for orthostatic hypotension (eg, fludrocortisone, midodrine) are not as effective as droxidopa (04).
Animal experiments with gene therapy have demonstrated full rescue of noradrenergic function in dopamine β-hydroxylase knockout mice, offering hope for an effective human gene therapy for this disorder in the future (10).
Surgery can correct the ptosis in affected individuals.
Treatment with dihydroxyphenylserine corrects or greatly ameliorates the orthostatic hypotension, improving functional performance and reducing the likelihood of accidental injury.
An uncomplicated cesarean section was performed at 36 weeks in a mother with dopamine beta-hydroxylase deficiency (52).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Douglas J Lanska MD MS MSPH
Dr. Lanska of the University of Wisconsin School of Medicine and Public Health has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neurogenetic Disorders
Jun. 01, 2026
Neurogenetic Disorders
May. 08, 2026
Neurogenetic Disorders
Apr. 30, 2026
Neuropharmacology & Neurotherapeutics
Apr. 23, 2026
Neurogenetic Disorders
Apr. 14, 2026
Neurogenetic Disorders
Apr. 14, 2026
Neurogenetic Disorders
Apr. 14, 2026
Neurogenetic Disorders
Apr. 14, 2026