






























Pyruvate dehydrogenase complex deficiency
May. 28, 2024
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Alpha- and beta-mannosidosis are autosomal recessive “storage” disorders associated with the excretion of mannose-rich oligosaccharides. Alpha-mannosidosis can present as either (1) a severe (infantile or type I) phenotype, with rapidly progressive mental retardation, hepatosplenomegaly, severe dysostosis multiplex, and often death between 3 and 12 years of age, or (2) a milder (juvenile-adult or type II) form of slowly progressive course, with survival into adulthood. Early hematopoietic stem cell transplantation provides significant benefit. Enzyme replacement therapy for alpha-mannosidosis has been developed and received approval in Europe. Glial fibrillary acidic protein, oligosaccharides, and other neuronal biomarkers are elevated in the CSF of most patients. Beta-mannosidosis is rarer and has a wide range of phenotypes, including (1) severe epilepsy, intellectual disability, hearing loss, quadriplegia, and early death; (2) spinocerebellar atrophy; and (3) isolated angiokeratomas.
|
• Alpha-mannosidosis can present as either (1) a severe (infantile or type I) phenotype, with rapidly progressive mental retardation, hepatosplenomegaly, severe dysostosis multiplex, and often death between 3 and 12 years of age or (2) a milder (juvenile-adult or type II) form with a slowly progressive course and survival into adulthood. | |
|
• Alpha-mannosidosis is characterized by facial dysmorphism, mental retardation, cataracts, corneal opacity, and hearing loss. | |
|
• Hearing impairment is an early sign of alpha-mannosidosis in children with a mild phenotype. | |
|
• Correct diagnosis requires measurement of enzyme activity in serum, plasma, or cells. | |
|
• Important biomarkers can be identified in most patients, which include elevated mannose complexes in MR spectroscopy and elevated oligosaccharides, GFAP, and biomarkers of neurodegeneration in CSF. | |
|
• Alpha-mannosidosis can be treated by early hematopoietic stem cell transplantation and enzyme replacement therapy. | |
|
• Enzyme replacement therapy for alpha-mannosidosis has been developed and received approval in Europe. | |
|
• Beta-mannosidosis is rare and has a wide range of phenotypes, including (1) severe epilepsy, intellectual disability, hearing loss, quadriplegia, and early death; (2) spinocerebellar atrophy; and (3) isolated angiokeratomas. |
Two distinct genotypes are included under the heading of mannosidosis: alpha-mannosidosis and beta-mannosidosis. Both are rare, and the phenotype is variable for both genotypes. The two forms of mannosidosis are both oligosaccharide lysosomal storage diseases and present as a wide range of phenotypes, in general being much slower in progression than the related lipidoses or mucopolysaccharidoses. Both mammalian genes have now been cloned, and the nature of the human mutations is actively being elucidated (93). Animal models are being used for therapeutic endeavors involving bone marrow transplants and gene therapy.
Alpha-mannosidosis. Alpha-mannosidosis was first described by Per-Arne Ockerman (1933-) in 1967 in Sweden; the proband was a mildly intellectually disabled child with a coarse body and face (69; 70). Because the coarse facial features resembled those in Hurler syndrome, alpha-mannosidosis was originally classified as a mucopolysaccharidosis but was later shown to be a distinct entity (69). In 1973, Ockerman described three additional cases in boys, aged 4, 5, and 10 years, with delayed psychomotor development, coarse facies, osteoporotic long bones with poor trabeculation and somewhat thick calvaria, and recurrent infections (03). This is the most typical clinical presentation (09).
Neurodegenerative alpha-mannosidosis in cattle and cats was subsequently described (39; 88). Obligate carrier cattle were shown to have 50% of normal alpha-mannosidase activity, verifying the disorder as a genetic rather than a toxic defect. Nevertheless, symptoms of affected cattle resemble those of normal North American cattle that had ingested "loco weed" (ie, certain species of Astragalus and Oxytropis): unstable gait, paralysis of the hind limbs, and head and neck tremor (90). The neurotoxic component of loco weed is an indolizidine alkaloid, "swainsonine," which is an inhibitor of alpha-mannosidases, including the lysosomal alpha-mannosidase involved in mannosidosis (90). Similar intoxication in goats was caused by Sida carpinifolia (24).
Beta-mannosidosis. Animals with beta-mannosidosis, first described in Nubian goats in 1981 (41) and subsequently in cattle (15), develop extensive central nervous system hypomyelination and hypothyroidism, which is manifest by tremor, ataxia, and early death (40; 74). Affected animals die in the neonatal period if intensive care is not provided.
Human beta-mannosidosis is less severe than beta-mannosidosis in goats or cattle. The first human case of beta-mannosidosis, described in 1986, was caused by a combined deficiency of beta-mannosidase and sulfamidase, leading to intellectual disability, coarse facial features, and mild skeletal changes reminiscent of the mucopolysaccharidoses (91). In 1988, Cooper and colleagues described two mildly intellectually disabled brothers, aged 29 and 44 years, with normal facies and no skeletal abnormalities; they had totally deficient beta-mannosidase activity and normal activity of other lysosomal hydrolases, including sulfamidase (17). Most subsequent cases of beta-mannosidosis have shown mild but progressive mental degeneration; however, there is a report of severely affected siblings with seizures in early childhood. Thus, diagnosis of beta-mannosidosis should be considered in mildly intellectually disabled children with progressive neurodegeneration and either mild skeletal abnormalities or mild hepatosplenomegaly.
Alpha-mannosidosis. Alpha-mannosidosis is a lysosomal storage disease that has been described in humans and various other mammals (eg, cattle, domestic cats, guinea pigs, and mice) (10). In humans, alpha-mannosidosis manifests as dysostosis multiplex (progressive facial and skeletal abnormalities), intellectual disability, motor impairment, and often sensorineural deafness, hepatosplenomegaly, and immunodeficiency with recurrent infections (10).
The index case was a boy who presented at birth as cyanotic and hypotonic with a weak cry but soon recovered and appeared normal until 12 months, by which time he had developed mild psychomotor delay and mild hepatosplenomegaly (69; 70). He became susceptible to infections (with hypogammaglobulinemia), and by 2 years of age, his growth slowed, and he developed lenticular abnormalities, macroglossia, dysmorphic features (eg, flat nose, large clumsy ears, and widely spaced teeth), and a lumbar gibbus (kyphosis). By age 4 years, he seemed uninterested in his surroundings, could not sit without support, could not grasp objects, and had hypotonic muscles. Plain x-rays showed skeletal abnormalities. He died at 4 years of age, following repeated attacks of violent restlessness, screaming, scratching, and vomiting.
Ockerman and colleagues described five additional cases with similar symptoms, including dysmorphic features, progressive psychomotor retardation, lumbar gibbus (ie, a form of structural kyphosis, where one or more adjacent vertebrae become wedged), corneal opacity with cloudy areas in the capsule of the lens, and hepatosplenomegaly but with longer life spans (ie, still alive at the age of 10 years) (03; 71).
Among 60 cases of alpha-mannosidosis, the clinical findings included the following (39):
|
Very common (more than two thirds of patients) | ||
|
• Dysmorphic face | ||
|
Moderately common (between one third and two thirds of patients) | ||
|
• Recurrent infections | ||
|
Fairly common (between one quarter and one third of patients) | ||
|
• Lens or corneal opacities | ||
Attempts to categorize alpha-mannosidosis cases into "classic" type I (severe) and type II (mild) have not been entirely successful due to the wide spectrum of manifestations, the variable features, and considerable overlap (03; 23; 66; 22; 57). Type I was associated with hepatomegaly and early death following severe infections, whereas type II was associated with hearing loss, mental retardation, and survival into adulthood (57). The range of manifestations in type II cases also includes slurred and slow speech with scanning, muscle flaccidity, tremor, equilibrium disturbances, and a deforming arthropathy (03; 22).
An alternate categorization of cases into three clinical types--mild (type I), moderate (type II), and severe (type III)--has been suggested, which is necessarily, albeit confusingly, numbered in reverse order from the prior dichotomy (16; 57; 58). Most individuals described fit into the moderate type by this categorization scheme.
|
• Type I (mild form): clinically recognized after 10 years of age, with a slowly progressive course but without skeletal abnormalities. | |
|
• Type II (moderate form): clinically recognized before 10 years of age, with slow progression and skeletal abnormalities. | |
|
• Type III (severe form): early onset, rapid progression, and skeletal abnormalities, leading to early death from primary CNS involvement or infection. |
Given the alternate categorization schemes, the use of type-x terminology requires specification of the categorization scheme being used. In any case, with the already wide and expanding clinical spectrum, prior categorization schemes are likely to prove inadequate.
The dysmorphic features can include an enlarged head, short neck, rounded eyebrows, saddle nose, and prominent forehead (57).
Skeletal abnormalities include vertebral abnormalities with kyphoscoliosis and coxarthrosis (57).
(A) Kyphoscoliosis with skeletal abnormalities of all vertebrae. (B) Orthopedic correction of kyphosis. (Source: Malm D, Nilssen O. Alpha-mannosidosis. Orphanet J Rare Dis 2008;3:21. Creative Commons Attribution License. http:/...
In cross-sectional investigations of 35 patients studied as part of two clinical trials of enzyme replacement therapy, intellectual disabilities were found in all patients, with IQ ranging from 30 to 81 (13; 10).
Milder patients typically present during adolescence with speech and hearing disorders and neurologic manifestations. Children with a mild phenotype can present with syndromic hearing impairment (48); neuropsychological evaluations found preserved visuospatial abilities but impaired language, which was attributed to hearing difficulties. Parenchymal lung disease may produce obstructive or restrictive impairment with air trapping (68). Immunocompromised patients often die before adulthood.
For cases with identified hearing impairment (at least three quarters of patients), the onset of deafness was noted in the first decade of life (38). Hearing impairment was sensorineural, of cochlear origin, bilateral, of a moderate degree (mean loss 62.76 dB; median 60 dB, standard deviation 12.5 dB), symmetrical, and stable over time. Audiometric curves have been reported as slightly sloping towards the higher tested frequencies, with a marked improvement at 4 kHz.
Beta-mannosidosis. Beta-mannosidosis is a rare lysosomal storage disease that can affect humans as well as other mammals (eg, cattle, goats, and Persian cats) (49; 02; 86; 80). Age of onset ranges from infancy to adulthood. The clinical presentation typically involves varying degrees of facial deformity, mental retardation, speech disorder, hearing loss, and recurrent respiratory infections (36). The disease is heterogeneous, even within the same family and even among patients with null mutations (05). Symptoms of the human disease are milder than those in animals with the same metabolic defect (02; 05).
Some patients present with seizures before 3 years of age. One patient presented with neonatal hypotonia with feeding difficulties and later suffered recurrent respiratory problems, esophageal dysmotility, and intellectual disability (28). A 14-year-old boy, presenting with bilateral thenar and hypothenar amyotrophy, had a demyelinating peripheral neuropathy (50). Two brothers, aged 29 and 44 years, had mild mental retardation with Fabry-like angiokeratomas but without dysmorphic features (17).
In another family, the proband was a girl who presented with psychomotor regression, dysmorphic features, bone deformities, and recurrent skin and respiratory infections, ultimately dying of bronchopneumonia at age 20 years (42); her elder brother had total beta-mannosidase deficiency with mild intellectual disability, mildly dysmorphic features, hearing impairment, and recurrent infections. Other patients have been reported with atypical Gilles de la Tourette syndrome (82), “pure” spinocerebellar degeneration (47), and hypomyelination (78).
An ethnic-specific variant of beta-mannosidosis, due to a homozygous c.2158-2A>G MANBA variant, is an important and previously unknown cause of hearing loss and intellectual disability among Central European Roma in the Czech and Slovak Republics (80).
The square symbol is for men, the circle for women. Clinically affected members with hearing loss are filled-in black. The number inside the symbols represents the number of siblings with that sex and clinical status. * - indic...
A 26-year-old woman (proband II/1 from a Slovak family) with hereditary hearing loss and beta-mannosidosis. The dashed line represents the Auditory Steady-State Response (ASSR), and objective estimation of the hearing threshold...
MANBA variant c.2158-2A>G screening among 345 anonymized normal hearing controls from Roma populations revealed a carrier/heterozygote frequency of 3.8%, three orders of magnitude higher than the frequency of this variant in the Genome Aggregation Database (gnomAD) (80).
Although small case series have suggested a slowly worsening progressive outcome over decades (07), two relatively large studies of alpha-mannosidosis, one a 12-month follow-up clinical evaluation and another based on family questionnaires, showed no convincing evidence of disease progression over time (04; 59). For example, the 6-minute walk test was only slightly worse in patients over 18 years of age compared to younger patients, and no age group showed significant change in distance over 1 year of follow-up. Nevertheless, motor test results correlated with levels of urinary oligosaccharides in the same patients. A historical review of 111 patients showed that high residual enzyme activity and later disease onset were associated with longer survival (97). In untreated patients with alpha-mannosidosis, pneumonia has been the primary cause of death during recent decades, followed by cancer (35).
In a study of long-term outcomes among 16 cases of alpha-mannosidosis, hearing loss was the primary manifestation found in 44%, followed by speech delay in 38% (43). On clinical follow-up, 88% experienced recurrent infections (mainly in the upper respiratory tract) and 75% required the use of a hearing aid. Hepatomegaly was found in six (of 13 who received abdominal ultrasonography); mitral valve prolapse was found in two (of 12 who underwent echocardiography). Delayed language development was observed in 56%, intellectual disability in 50%, and hypertonicity in 6%.
The prognosis of beta-mannosidosis is poor with progressive and unrelenting degeneration, although the symptoms may be mild enough to permit life beyond adolescence.
A 12-year-old boy presented with progressive gait unsteadiness, lower limb weakness, and cognitive problems (47). Early psychomotor development was normal, but by 4 years of age, mental slowness was noted at school. Clinical examination showed impaired cognitive function, a symmetrical spastic tetraparesis with bilateral Babinski signs, and cerebellar ataxia. There were no skin lesions or skeletal deformities.
Sensory evoked potentials elicited no cortical responses. Visual evoked potentials showed increased latencies of the P100 potentials (117 ms on the right and 120 ms on the left). Auditory evoked potentials revealed a wave V/I amplitude ratio (an index to adjust for intersubject variability in wave amplitudes) less than 50% in the presence of normal latencies, suggesting central dysfunction in the upper brainstem. Electromyography and nerve conduction studies were normal. At 12 years of age, cranial CT showed diffuse cortical and subcortical atrophy. At age 15 years, cranial MRI showed diffuse cortical and subcortical atrophy with predominant involvement of the brainstem and the vermis, without evident white matter changes. Serial cranial MRI studies (at ages 18, 22, and 25 years) showed no significant change. At age 18 years, beta-mannosidase activity in peripheral blood leukocytes was severely decreased to 4 nmol/h/mg protein (normal, 71 nmol/h/mg protein), with a similar decrease in plasma.
Sequencing all exons (and their flanking regions) of the MANBA gene showed that the patient was homozygous for the novel mutation c.1922G>A, replacing arginine 641 with histidine (p.R641H) in exon 14. Heterozygosity for the same mutation was confirmed in both parents. They had intermediate levels of leukocyte beta-mannosidase activity (mother: 35 nmol/h/mg protein, father: 42 nmol/h/mg protein). Of the three siblings, heterozygous mutations were found in the patient’s 19-year-old brother and 26-year-old sister; both were asymptomatic and had mildly decreased beta-mannosidase activity levels (68 and 51 nmol/h/mg protein, respectively). The remaining brother had a normal beta-mannosidase activity level and did not carry the gene mutation.
Symptoms progressively worsened. At age 18 years, his IQ was 60. By age 26 years, he was bedridden with spastic tetraplegia and pseudobulbar syndrome (severe dysphagia and spastic dysarthria).
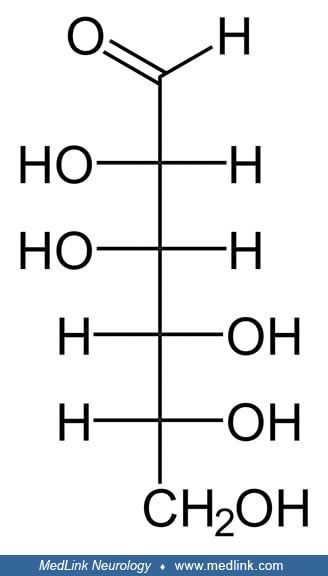
Mannose. D-Mannose is a sugar monomer of the aldohexose series of carbohydrates. Five of the carbons have one hydroxyl functional group (–OH), and one has a carbonyl group (C=O). When the carbonyl of a 6-carbon sugar molecule is on one end of the carbon chain, forming an aldehyde (ie, a formyl group [–CH=O]), as is the case for mannose, the sugar is called an aldohexose. Or, more generically, it is an aldose, a monosaccharide (a simple sugar) with a carbon backbone chain with a carbonyl group on the endmost carbon atom (ie, an aldehyde) and hydroxyl groups on all of the other carbon atoms. The carbons are numbered 1 to 6 starting at the carbonyl end.
D-mannose is a C-2 epimer of D-glucose. An epimer is an isomer with a different configuration of atoms around one of several asymmetric carbon atoms (ie, chiral centers) present. Consequently, D-mannose and D-glucose are also diastereomers (ie, non-mirror image, non-identical stereoisomers).
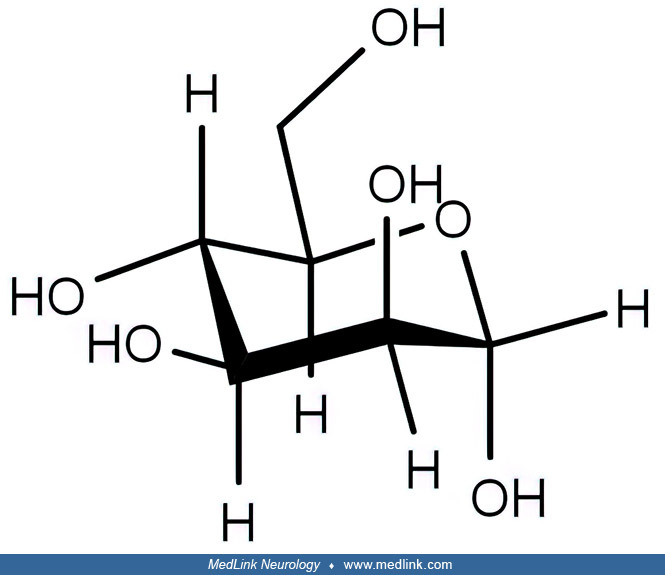
Mannose, like other hexoses, exists in open-chain and cyclic isomeric forms that easily interconvert in aqueous solutions, with the cyclic form being highly preferred.
The open-chain form has four chiral centers, whereas the cyclic forms have five chiral centers. Mannose has two different-sized ring forms, the furanose (five-membered) form and the pyranose (six-membered) form. Each type of ring can have either an alpha or beta configuration at the anomeric position (ie, the carbon, C-1, that bears the aldehyde functional group in the sugar's open-chain form), yielding alpha-D-mannofuranose, beta-D-mannofuranose, alpha-D-mannopyranose, and beta-D-mannopyranose.
The commonly employed Haworth projection of the ring forms assumes that the ring is a planar structure, but for the pyranose (six-membered) ring forms, the ring is not planar but instead has a "chair" three-dimensional configuration.
Mannose metabolism. The digestion of many polysaccharides and glycoproteins yields mannose, which is phosphorylated by hexokinase to generate mannose-6-phosphate. Mannose-6-phosphate is converted to fructose-6-phosphate by the enzyme phosphomannose isomerase and then enters the glycolytic pathway or is converted to glucose-6-phosphate by the gluconeogenic pathway of hepatocytes.
Glycosylation. Glycans are polysaccharides (ie, multiple monosaccharides linked through O-glycosidic linkages) (37), but in practice, the term glycan may refer to the carbohydrate portion of a glycoconjugate (eg, glycoprotein, glycolipid, or a proteoglycan), even if the carbohydrate is only an oligosaccharide (25).
Post-translational glycan modifications of proteins are important for many purposes within the cell, including inter alia protein folding, protein stabilization, steric protection from proteolytic degradation, regulation of protein-protein interactions, and cell-to-cell adhesion (60). Up to 70% of mammalian proteins are glycosylated.
The glycans are usually attached to proteins via “N” or “O” linkages, with N-glycosylations being most common. N-glycans are attached to the amide nitrogen (ie, the "N" linkage) of an asparagine (Asn) residue on proteins, whereas O-glycans are attached to the hydroxyl group (the “O” linkage) of either serine (Ser) or threonine (Thr). Rarely, glycosylation involves C-linked glycans, where a glycan is added to a carbon (the “C” linkage) on a tryptophan side-chain.
Mannose is a dominant monosaccharide in N-linked glycosylation. The synthesis of an N-linked glycan begins in the endoplasmic reticulum, continues in the Golgi, and ends at the plasma membrane, where the finished glycoproteins are either secreted or become embedded in the plasma membrane (60).
N-glycans are attached to proteins at specific motifs: asparagine-X-xerine or asparagine-X-threonine, where X is any amino acid except proline. During the process of N-glycosylation, monosaccharides (often donated by nucleoside sugars, such as uridine diphosphate mannose [UDP-mannose] and guanosine diphosphate mannose [GDP-mannose]) are sequentially added to the glycan structure.
Initially, two N-acetylglucosamine (NAc) residues are added consecutively to dolichol phosphate (DOL) in the cytosol, followed by the addition of several mannose (Man) residues. After formation of the intermediate Man5HexNAc2-PP-Dol, the complex is flipped into the endoplasmic reticulum lumen, where further mannose residues and then glucose (Glc) residues are added, donated by Glc-P-dolichol. The Glc3Man9GlcNAc2-PP-dolichol precursor glycan is then transferred to an asparagine (Asn) residue on a newly synthesized protein. Glycosidases and glycosyltransferases then modify the precursor glycan to generate thousands of unique structures, which can be separated into three very broad structural categories: high mannose, hybrid, and complex.
Typically, mature human glycoproteins are complex and only contain three mannose residues buried under sequential modification by GlcNAc, galactose, and sialic acid. This is important as the innate immune system in mammals is programmed to recognize exposed mannose residues as foreign.
Glycoprotein catabolism. During normal cellular catabolism, mature glycoproteins enter the lysosomal degradation pathway and are digested by proteinases and glycosidases within lysosomes. These enzymes degrade glycoproteins into amino acids and monosaccharide fragments that can either be excreted or transported to the cytosol for reuse. Free monosaccharides derived from lysosomal degradation enter the recycling or salvage pathway of monosaccharides and constitute an important source for glycans in the endoplasmic reticulum and Golgi apparatus. When these hydrolases are absent or deficient, undigested material accumulates in lysosomes and disrupts cellular functions.
Mannosidosis is a group of inborn errors of metabolism, usually due to a deficiency of lysosomal alpha-mannosidase or beta-mannosidase (46).
Alpha-mannosidosis. Alpha-mannosidosis is transmitted as an autosomal recessive trait.
The global carrier frequency of alpha-mannosidosis in a global genomic database (Genome Aggregation Database) comprising a collection of 125,748 exomes was 0.23%; the highest carrier frequency was observed in the Finnish at 0.49%, and East Asians had the second highest carrier frequency at 0.30% (72). Globally, the approximate incidence of alpha-mannosidosis was calculated at 1 in 784,535, 1 in 166,801 Europeans (Finnish), and 1 in 431,689 East Asians. By integrating the data from the 8936 Koreans in this and other databases, the carrier frequency of alpha- mannosidosis in the Korean population was 0.04%, and the estimated incidence was 1 in 19,963,024.
Carrier frequency and estimated incidence of alpha-mannosidosis according to population in a global genomic database (Genome Aggregation Database) that comprises a collection of 125,748 exomes. Circle size: carrier frequency of...
The inherited deficiency of acid alpha-mannosidase (EC 3.2.1.24), due to a mutation in the MAN2B1 gene (OMIM 609458) on chromosome 19p13.13, leads to a failure in the catabolism of glycoproteins and the accumulation of a range of manno-oligosaccharides in hypertrophied lysosomes in all tissues, including brain and liver. The numerous storage vacuoles in the liver, as seen on electron microscopy, appear as finely granular material dispersed on an electron-lucent background. Neutrophils and lymphocytes in the peripheral blood and marrow are typically vacuolated and contain coarse, dark granules, but this is not diagnostic (57).
Electron micrograph of (A) a vacuolated lymphocyte from a patient with alpha-mannosidosis, and (B) a lymphocyte from a normal control. (Source: Malm D, Nilssen O. Alpha-mannosidosis. Orphanet J Rare Dis 2008;3:21. Creative Comm...
The MAN2B1 gene has been cloned, shown to code a protein of 961 amino acids, and localized to chromosome 19q (GenBank RefSeq NG 008318.1) (65; 51). Many different human mutations have been reported (67; 79).
Alpha-mannosidases can be divided into two groups: class 1 can hydrolyse only (1 --> 2) linkages (eg, Golgi mannosidase I), whereas class 2 can hydrolyse (1 --> 2), (1 --> 3), and (1 --> 6) linkages (eg, lysosomal alpha-mannosidase) (20). Class I alpha-mannosidases derived from an enzyme of the endoplasmic reticulum involved in the degradation of glycans on incompletely assembled or misfolded glycoproteins, whereas Class II alpha-mannosidases derived from a lysosomal enzyme responsible for turnover. Subsequent alpha-mannosidases with new processing or catabolic functions developed from these by loss or gain of critical insertion or retention sequences.
Human alpha-mannosidase has been purified and characterized (57). Enzyme deficiency causes the accumulation of free oligosaccharides and over-glycosylation of proteins (18).
A modest genotype-phenotype association has been demonstrated for alpha-mannosidosis type II. Among 66 patients with alpha-mannosidosis type II, the MAN2B1 genotype and the subcellular localization of mutant MAN2B1 in cultured cells were associated with cognitive function, upper limb coordination, balance, forced vital capacity, and the CSF storage of oligosaccharides, although there was considerable overlap across groups for all of the comparisons (12). Patients with missense mutations and enzyme localization in the lysosome had better neurocognitive function and lower concentrations of oligosaccharides in the CSF compared to those with missense mutations and protein localization in the endoplasmic reticulum or those with nonsense mutations (12).
The clinical variability among those sharing the same MAN2B1 genotype suggests that other genes interact to produce the phenotypic variation. To explain the intrafamilial variability of cognitive impairment as a feature of alpha-mannosidosis in two consanguineous Tunisian families, exome analysis identified two likely pathogenic variants in GHR (which encodes a member of the type I cytokine receptor family that is a transmembrane receptor for growth hormone) and SLC19A3 (which encodes thiamine transporter 2, also known as solute carrier family 19 member 3) genes potentially associated with cognitive decline (62).
In a study of six patients, immunodeficiency was evident at both the humoral and cellular levels, manifested by lower levels of antibody in postimmunization serum and low intracellular bacteria-killing capacity (56).
Beta-mannosidosis. Beta-mannosidosis is transmitted as an autosomal recessive trait.
The inherited deficiency of acid beta-mannosidase, due to a mutation in the MANBA gene (OMIM 609489) on chromosome 4q24, leads to a failure in the catabolism of glycoproteins and the predominant accumulation of a manno-disaccharide in humans. In goats and cows, it leads to a manno-trisaccharide in hypertrophied lysosomes in all tissues, including the brain and liver. These can readily be seen on electron microscopic examination as large, clear inclusion bodies. Foamy histiocytes and macrophages containing periodic acid-Schiff-positive, diastase-resistant material are commonly seen.
The human MANBA gene has been cloned (02), and the coding sequence on chromosome 4 encodes a protein of 879 amino acids. Mutation analysis of a Czech Romani (gypsy) family with two siblings differently affected with beta-mannosidosis demonstrated a homozygous A-to-G transition two base pairs upstream of a splice acceptor site, causing exon skipping and abnormal mRNA splicing (02).
Mannosidosis immunodeficiency syndrome. A single case has been reported with combined immune deficiency harboring biallelic mutations in the mannosidase alpha class 2B member 2 (MAN2B2) gene, affecting both N-glycan synthesis and glycan degradation (87). MAN2B2 is a core-specific alpha-mannosidase (OMIM 618899) that cleaves the alpha-1,6 mannose linkage of the N-glycan core structure following cleavage of a GlcNAc residue from the reducing terminus.
The patient was an 8-year-old girl of consanguineous healthy parents from Saudi Arabia. As a neonate, she suffered from recurrent pneumonia and thrush. She manifested small-vessel vasculitis affecting her distal extremities and ears starting about age 3 months, polyarthritis starting at age 9 months, and a thrombotic stroke with left hemiparesis around age 16 months. She also developed severe chronic diarrhea, recurrent oral herpes, severe respiratory infections requiring intubation, severe failure to thrive, and psychomotor developmental delay. At age 4 years, she was microcephalic with growth retardation, speech delay, strabismus, a beaked nose, hyperextensible skin on the dorsum of her hands, pectus carinatum, and mild hepatomegaly. She could walk only with support. Cranial MRI showed a large chronic infarct of the right frontoparietal region with encephalomalacia, gliosis, areas of cerebrocortical laminar necrosis and hemosiderin deposition, and evidence of right midbrain Wallerian degeneration. Whole-exome sequencing identified a homozygous missense variant in the MAN2B2 gene (Chr4:g.6575322G> A; p.Asp38Asn). Abnormalities of glycosylation and lysosomal involvement were reversed in vitro on lentivirus-mediated transfer of wild-type MAN2B2. At 5 years of age, the girl received hematopoietic stem cell transplantation from her phenotypically HLA-matched father. Six months after hematopoietic stem cell transplantation, her immunoglobulin production and T- cell count and function all improved, with stabilization of the disease and resolution of infections.
Alpha-mannosidosis and beta-mannosidosis are both rare disorders (1 in 100,000 live births) but are underrecognized and, hence, underdiagnosed (61; 92).
The single nucleotide polymorphism located in the 3′UTR of mannosidase beta, rs7665090, is associated with multiple sclerosis susceptibility (27). The rs7665090*GG genotype caused a significant beta-mannosidase enzymatic deficiency correlated with lysosomal dysfunction as well as decreased metabolic activation in lymphocytes of patients with multiple sclerosis compared to those of rs7665090*GG controls.
Prenatal diagnosis is available.
Speech and hearing abnormalities in a mildly intellectually disabled child may help distinguish mannosidosis from other lysosomal storage diseases. Early respiratory problems may also be of some diagnostic value. Neither alpha-mannosidosis nor beta-mannosidosis is characterized by overt hepatosplenomegaly. Symptoms of mannosidosis are generally milder than those seen in fucosidosis. Biochemical tests are essential to distinguish alpha-mannosidosis, beta-mannosidosis, alpha-fucosidosis, and aspartylglucosaminuria.
Previously, a first step in the laboratory diagnosis of oligosaccharidosis was to demonstrate abnormal oligosaccharides in urine, usually by the poorly sensitive thin layer chromatography method (20), but liquid chromatography with tandem mass spectrometry (LC/MS/MS) is more specific and much more sensitive than thin layer chromatography and is applicable to both urine and amniotic fluid for alpha- and beta-mannosidosis (76; 45; 84).
However, there is a move to the determination of enzymatic activity as the first choice for screening and confirmatory diagnosis, using either leukocytes or dried blood spots (although a result based on dried blood spots should be confirmed in leucocytes or cultured skin fibroblasts) (58; 32). Molecular genetic testing (ie, testing based on DNA or RNA sequencing) of the patient and his or her parents should be performed as a confirmatory step and for family investigations (58; 32). Some countries use next-generation sequencing in either targeted gene panels for specific clinical manifestations or exome or whole-genome sequencing (32). Future diagnostics will focus on molecular genetic testing as the initial approach, with determination of enzyme activity or oligosaccharide levels being done as biochemical confirmation of molecular results (32).
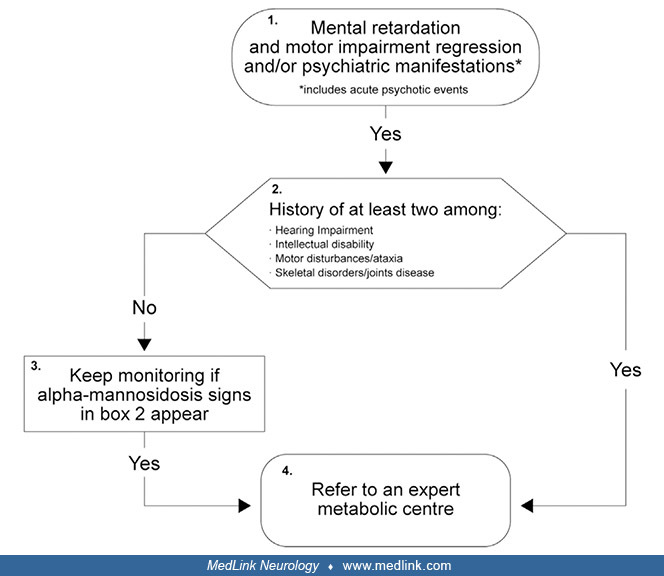
Alpha-mannosidosis. Diagnosis of alpha-mannosidosis is often delayed, in part because of its rarity, gradual onset, and heterogeneity of presentation, but also because many signs and symptoms of the disease overlap with those of other lysosomal diseases. A clinical diagnostic algorithm to facilitate early recognition and diagnosis of alpha-mannosidosis has been developed, with different flowcharts applicable to suspect cases of patients either under 10 years of age or 10 years and older (32).
MPS = mucopolysaccharidosis. (Source: Guffon N, Tylki-Szymanska A, Borgwardt L, et al. Recognition of alpha-mannosidosis in paediatric and adult patients: presentation of a diagnostic algorithm from an international working gro...
(Source: Guffon N, Tylki-Szymanska A, Borgwardt L, et al. Recognition of alpha-mannosidosis in paediatric and adult patients: presentation of a diagnostic algorithm from an international working group. Mol Genet Metab 2019;126(...
The approach focuses on recognizing the clinical features that, if met, warrant referral to an "expert metabolic center" for laboratory diagnosis and management (32).
Serum or plasma can be used to demonstrate a profound (0% to 15%) deficiency of the ability to hydrolyze 4-methylumbelliferyl-alpha-D-mannopyranoside at pH 4.0 in a sodium acetate buffer. Leukocytes, cultured fibroblasts, amniotic fluid cells, and tissue biopsy samples have all been used to give an unambiguous diagnosis based on the demonstration of deficient alpha-mannosidase activity, whereas the activities of other lysosomal enzymes are either normal or elevated. Invasive procedures are not required for diagnosis; moreover, analysis of enzyme activity in liver, for example, may be problematic because of the presence of a neutral beta-mannosidase that is unaffected in the disease.
In alpha-mannosidosis, MRI shows cerebral and cerebellar atrophy with ventriculomegally and diffuse symmetric dysmyelination (29; 54; 43). Other findings may include cranial abnormalities (eg, brachycephaly, trigonocephaly), marked thickening of the calvarium, inferior vermis hypoplasia, fluid intensities in optic nerve sheaths, an enlarged cerebellar medullary cistern, a tight foramen magnum associated with a cervical syrinx, and hyperintensity of the thalami and dentate nuclei (73; 29; 55; 43).
Cranial MRI (T1-weighted) showing a large cisterna magna and mild brachycephalic appearance in a boy with mannosidosis. Symptom onset was at 30 months, and diagnosis was at 62 months. (Source: Kose E, Kasapkara ÇS, İnci A, et a...
Cranial MRI (T1-weighted) showing fluid intensities in optic nerve sheaths, thickening at calvarial diploic spaces in a boy with mannosidosis. Symptom onset was at 36 months, and diagnosis was at 86 months. (Source: Kose E, Kas...
Cranial MRI (T1-weighted) showing a mild brachycephalic appearance, inferior vermis hypoplasia, and a large cisterna magna. Symptom onset was at 32 months, and diagnosis was at 148 months. (Source: Kose E, Kasapkara ÇS, İnci A,...
Beta-mannosidosis. Serum or plasma can be used to show a profound (0% to 10%) deficiency of the ability to hydrolyze 4-methylumbelliferyl-b-D-mannopyranoside at pH 4.0 in a sodium acetate buffer. Leukocytes, cultured fibroblasts, amniotic fluid cells, and tissue biopsy samples have all been used to give an unambiguous diagnosis based on the demonstration of deficient activity of beta-mannosidase, whereas the activities of other lysosomal enzymes are either normal or elevated (41). Invasive procedures are not required for diagnosis.
Until recently, treatment of mannosidosis has been largely symptomatic.
Enzyme replacement therapy. Human lysosomal alpha-mannosidase cDNA was cloned and expressed in Chinese hamster ovary cells. Recombinant alpha-mannosidase was shown to be taken up by cultured alpha-mannosidosis fibroblasts and trafficked to lysosomes via the mannose 6-phosphate pathway where it reduced the amounts of stored mannose-containing oligosaccharides (06).
A human trial of recombinant human lysosomal acid alpha-mannosidase (velmanase alfa) in 33 patients with alpha-mannosidosis demonstrated a reduction in serum oligosaccharide levels and clinical improvement in motor function (ie, on the 3-minute stair climb test), endurance, and pulmonary function (52; 01; 64). Such clinical benefits may confer improved health-related quality of life (52). The European Medicine Agency approved recombinant enzyme (velmanase alfa) therapy for the non-neurologic manifestations of the disease, based largely on improved walking ability of affected patients (11; 34; 52; 58). Long-term safety and efficacy appear to be good in small series: most adverse effects are mild or moderate, although four of five cases in one study developed antidrug antibodies (three were neutralizing), and all children improved in one or more efficacy assessments (serum oligosaccharide concentrations, immunological profile, hearing, and quality of life) (31).
Although there have been no direct comparisons of the effectiveness of enzyme replacement therapy and hematopoietic cell transplant in alpha-mannosidosis, enzyme replacement therapy has at best only limited blood-brain barrier penetration; consequently, hematopoietic cell transplant is the modality thought to have the best potential for neurocognition preservation (64). However, in experiments with animal models of the disease, high-dose enzyme infusions reduced oligosaccharide storage in the brain and partially corrected cognitive function, suggesting that some enzymes may cross the blood-brain barrier through a process independent from the mannose-6-phosphate receptor that normally provides that capacity in early postnatal life (08; 19; 85; 26).
Hematopoietic stem cell transplant. The apparently successful bone marrow transplant in a cat with alpha-mannosidosis provided support to pursue stem-cell transplantation in humans (89); enzyme was detected in neurons and glia following transplant, and the cat showed no evidence of the progressive degenerative disease seen in untreated, affected littermates.
Hematopoietic stem cell transplant can attenuate CNS disease and is now a therapeutic option for human alpha-mannosidosis, but it should be performed as early as possible to minimize the accumulation of storage material and the resulting irreversible worsening of neurologic function and skeletal development (44; 75; 30; 14; 63; 94; 64). Because early hematopoietic stem cell transplant with complete engraftment can reverse or prevent most of the clinical manifestations of alpha-mannosidosis, early transplantation is likely to yield better results than transplantation performed once symptoms and signs are fully declared (14; 94).
High-field in vivo proton magnetic resonance spectroscopy after bone marrow transplantation demonstrates the disappearance of the mannose-containing oligosaccharide resonance complex (21). Accumulated cerebral mannose-containing oligosaccharides were evident on magnetic resonance spectroscopy in a patient who subsequently received a bone marrow transplant from a haploidentical noncarrier sibling; however, this pathologic storage material disappeared as early as 9.5 months after bone marrow transplantation and did not reaccumulate up to 5.5 years after bone marrow transplantation (21).
More than 20 alpha-mannosidosis patients have undergone hematopoietic stem cell transplant, more than any other glycoprotein disorder (63; 64). The 88% 5-year survival rate is comparable to those transplanted for Hurler disease or other nonmalignant diseases (63; 64). Peripheral blood alpha-mannosidase activity normalized in tested children; treated children also improved developmentally, with no regression in previously learned skills. None of the children who received transplants achieved normal hearing capacity, but some participants were able to discontinue hearing aids temporarily after transplant (63; 64).
Bridge enzyme replacement therapy can be used in the pre- and peri-transplant phases before hematopoietic stem cell transplantation, and such early combined intervention may reduce the disease progression and the urine and plasma content of mannosyl-oligosaccharides (81).
Intravascular gene therapy. The potential utility of gene therapy has been demonstrated in feline experiments with symptomatic alpha-mannosidosis cats, a homolog of the human disease (95; 96; 77).
In 2016, widespread improvement of clinical disease, survival, and neuropathology was achieved in a symptomatic alpha-mannosidosis cat using a single cisterna magna infusion of the adeno-associated virus vector AAV1 expressing the feline alpha-mannosidase gene (95). Unfortunately, AAV vectors are generally inadequate for treating the whole brain using noninvasive injection, including intravascular injection; the distribution of AAV in the CNS of large mammals has been restricted after systemic AAV injection so that gene transduction of cells is concentrated in the lower brain and spinal cord with relatively few cells transduced in the forebrain of large animal species (96).
The search for a vector that can efficiently cross the blood-brain barrier after intravascular injection led to the identification of the AAV vector AAVhu.32, which is in the same clade as serotype 9 of the adeno-associated virus (96). In 2020, high-dose delivery via the carotid artery in a symptomatic alpha-mannosidosis cat produced complete histological correction of the brain, improved clinical measures, and increased lifespan (96). Treatment response was dose-dependent, and intra-arterial injection was more effective than intravenous delivery.
Other treatments. Patients with type II mannosidosis and end-stage renal failure may benefit from renal transplantation; in one case, after 6 years of regular hemodialysis, urinary oligosaccharides normalized, and the clinical course stabilized for 3 years after renal transplantation (83).
Enzyme replacement therapy. There are no preclinical studies or clinical trials of enzyme replacement therapy in beta-mannosidosis (64).
Hematopoietic stem cell transplant. Hematopoietic stem cell transplant has been performed in only one patient with beta-mannosidosis (53). A 4-year-old boy was diagnosed with beta-mannosidosis after presenting with macrocephaly and delayed gross motor development (including deficits in balance and gait). Following hematopoietic stem cell transplant, the boy achieved engraftment, CSF and plasma enzyme levels nearly normalized, and urine oligosaccharides decreased dramatically. However, 2 years after the hematopoietic stem cell transplant, the boy had persistent impairment in motor function and a decline in visuomotor integration, memory, and IQ.
No specific published information has been found.
Detailed description of successful anesthesia for 10 alpha-mannosidosis patients undergoing 14 procedures was provided (33). Airway management should be based on individual, and a thorough pre-anesthesia airway assessment should be evaluated for each patient (33).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Douglas J Lanska MD MS MSPH
Dr. Lanska of the University of Wisconsin School of Medicine and Public Health and the Medical College of Wisconsin has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neurogenetic Disorders
May. 28, 2024
Neurogenetic Disorders
May. 15, 2024
Developmental Malformations
May. 15, 2024
Neurogenetic Disorders
May. 14, 2024
Neurogenetic Disorders
May. 14, 2024
Developmental Malformations
May. 14, 2024
Neurogenetic Disorders
May. 14, 2024
Neurogenetic Disorders
May. 14, 2024