Methylmalonic acidemia
Jun. 01, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
This article reviews classic Lesch-Nyhan disease and less severe Lesch-Nyhan disease variants. These metabolic disorders result from mutations in the HPRT1 gene, which encodes for the purine salvage enzyme hypoxanthine-guanine phosphoribosyltransferase (HPRT). Lesch-Nyhan disease is an X-linked genetic disorder characterized by hyperuricemia, intellectual disability, dysarthric speech, early-onset hypotonia, later-onset dystonic movement disorder, and compulsive self-mutilation accompanied by an extended cognitive and behavioral phenotype.
Management of Lesch-Nyhan disease incorporates psychosocial support to the patient and caregivers, behavioral and pharmacological treatment, use of protective equipment, and dental, orthopedic, and nephrology management. Goals of treatment in Lesch-Nyhan disease include reduction of self-mutilating and aggressive behavior, prevention of renal failure and urologic complications related to hyperuricemia, control of dystonic movements, and orthopedic correction of hip subluxation and dislocation.
|
• Lesch-Nyhan disease is a metabolic disorder resulting from deficiency of the enzyme hypoxanthine-guanine phosphoribosyltransferase that is involved in recycling the purines hypoxanthine and guanine. | |
|
• This X-linked genetic disorder is associated with hyperuricemia, intellectual disability, dysarthric speech, early hypotonia, dystonic movement disorder, and compulsive self-mutilation. | |
|
• Lesch-Nyhan disease is characterized by an extended behavioral, neurologic, and neurocognitive phenotype. | |
|
• Human studies and animal models of Lesch-Nyhan disease have documented dopaminergic dysfunction. | |
|
• Management of Lesch-Nyhan disease incorporates psychosocial support to the patient and caregivers, behavioral and pharmacological treatment, use of protective equipment, and dental, orthopedic, and nephrology management. | |
|
• Goals of treatment in Lesch-Nyhan disease include reduction of self-mutilating and aggressive behavior, prevention of renal failure and urologic complications related to hyperuricemia, control of dystonic movements, and orthopedic correction of hip subluxation and dislocation. |
Lesch-Nyhan disease was initially described in 1964 in two brothers by American cardiologist Michael Lesch (1939-2008) and his mentor, pediatrician and biochemical geneticist William Nyhan (b. 1928), both working at the Laboratory of General and Comparative Biochemistry at the NIH National Institute of Mental Health in Bethesda, Maryland (100; 120). The 4-year-old younger brother had been diagnosed with cerebral palsy and presented with hematuria. Both he and his 8-year-old brother had intellectual disability, a movement disorder with dystonia, renal stones, and self-biting.
In 1967, just 3 years after the clinical recognition of the disorder by Lesch and Nyhan, American rheumatologist and biochemical geneticist J(arvis) ("Jay") Edwin Seegmiller (1920-2006) and colleagues from the National Institutes of Health discovered the biochemical basis of Lesch-Nyhan syndrome, establishing it as a disease with a defined cause (153).
J(arvis) ("Jay") Seegmiller is best known for his role in discovering the biochemical basis of Lesch-Nyhan syndrome, establishing it as a disease with a defined cause. (Source: National Institutes of Health. See: Anonymous. Drs...
Hypoxanthine-guanine phosphoribosyltransferase (HPRT) was identified as the missing enzyme involved in this metabolic disorder.
Most patients with classical Lesch-Nyhan disease have low or undetectable levels of the HPRT, rarely more than 1%. The complete amino acid sequence for HPRT was described in 1982, and its three-dimensional structure was described using x-ray crystallography in 1994, along with the effects of single amino acid substitutions on the stability and activity (181; 40). Kinetic mechanisms of HPRT function were described in 1997 (187).
The gene was localized to the long arm of the X chromosome (q26-q27) in 1979, and the organization of the HPRT1 gene (approximately 44 kb; 9 exons) was described in 1986 (13; 94).
|
• Lesch-Nyhan disease includes the following features: hyperuricemia, intellectual disability (intellectual developmental disorder), dysarthric speech, hypotonia, dystonia, and compulsive self-injury (usually beginning with the eruption of teeth). | |
|
• Self-injury may begin as early as 1 year of age and occasionally as late as the teens. | |
|
• Self-injury is the characteristic feature of classic Lesch-Nyhan disease, but it is not characteristic of Lesch-Nyhan variants (ie, with HPRT enzyme activity of more than 2%). |
Lesch-Nyhan disease. Lesch-Nyhan disease includes the following features: intellectual disability (intellectual developmental disorder), dysarthric speech, hypotonia, dystonia, compulsive self-injury (usually beginning with the eruption of teeth), hyperuricemia, tophi, orange-colored and possibly gritty urine (eg, “orange sand” in the diapers of infants with this disorder from urate crystals), and renal dysfunction (100; 188; 58; 22; 108; 85; 95; 116; 158). At birth, infants with Lesch-Nyhan disease have no apparent neurologic dysfunction. Many affected children seem to develop normally for up to 6 months before developmental delay and neurologic signs become evident, but some children have evident manifestations before 3 months of age (eg, hypotonia, recurrent vomiting, and difficulty managing oral secretions). At approximately 6 to 12 months of age, extrapyramidal signs appear, primarily dystonia. Hypotonia and psychomotor retardation may occur in the first year of life, whereas other neurologic symptoms, such as choreoathetosis and self-destructive biting of the fingers and lips, usually appear between the ages of 2 and 4 years (158). Uncommonly, seizures may occur, although this is often in the setting of other complications, such as respiratory failure and severe respiratory acidosis.
In a cohort of 101 French and Italian patients with Lesch-Nyhan disease or variant cases, the median age of onset was 1.0 years for involuntary movements and 3 years for self-mutilation behavior (108). Renal manifestations occurred in two thirds of patients; renal manifestations occurred at any age with a median onset age of 1.1 years (108). Gout, which occurred in a quarter of patients, appeared later in the disease course (median onset age 18 years) and was more frequent in attenuated variants than in Lesch-Nyhan disease (108).
Tophi may appear in various places, including the external ear, fingers, toes, and ankles (06). Tophi may also spontaneously rupture and discharge tophaceous fragments.
(Source: Ambarsari CG, Cahyadi D, Sari L, et al. Late diagnosis of Lesch-Nyhan disease complicated with end-stage renal disease and tophi burst: a case report. Ren Fail 2020;42(1):113-21. Creative Commons Attribution License. h...
(Source: Ambarsari CG, Cahyadi D, Sari L, et al. Late diagnosis of Lesch-Nyhan disease complicated with end-stage renal disease and tophi burst: a case report. Ren Fail 2020;42(1):113-21. Creative Commons Attribution License. h...
(Source: Ambarsari CG, Cahyadi D, Sari L, et al. Late diagnosis of Lesch-Nyhan disease complicated with end-stage renal disease and tophi burst: a case report. Ren Fail 2020;42(1):113-21. Creative Commons Attribution License. h...
(Source: Ambarsari CG, Cahyadi D, Sari L, et al. Late diagnosis of Lesch-Nyhan disease complicated with end-stage renal disease and tophi burst: a case report. Ren Fail 2020;42(1):113-21. Creative Commons Attribution License. h...
In a 15-year-old boy with Lesch-Nyhan disease, a tophus of the left ankle burst and discharged stones. (Source: Ambarsari CG, Cahyadi D, Sari L, et al. Late diagnosis of Lesch-Nyhan disease complicated with end-stage renal dise...
Early reports of Lesch-Nyhan syndrome emphasized spasticity with pyramidal tract signs, choreoathetosis, compulsive self-injurious behavior, and mental handicap as the most characteristic features. Spasticity is a variable feature of Lesch-Nyhan disease. Some cases with spasticity result from secondary complications, including atlantoaxial instability, which can present with neck pain and be the harbinger of progressive quadriparesis or even quadriplegia (157). Later studies noted additional features, including extrapyramidal features characteristic of dysfunction of the motor circuits of the basal ganglia and particularly a dystonic movement disorder (178; 173; 91). In a study of 44 patients ranging in age from 2 to 38 years, the movement disorder was characterized as a severe action dystonia superimposed on baseline hypotonia (91). Other movement disorders also occur, including hemiballismus, blepharospasm, and ocular tics (90). Associated disturbances of ocular motility, cognition, and behavioral control may reflect disruption of other basal ganglia circuits, especially because neurochemical studies have demonstrated 60% to 90% reductions in the dopamine content of the basal ganglia (173).
Intellectual disability is usually in the mild-to-moderate range, although some individuals test in the low-normal range of intelligence. Although affected individuals generally continue to acquire new information and skills over time, their standardized test scores decline, reflecting increasing separation from their age-matched peers (110; 111). Deficits in working memory are common, particularly on tasks that involve considering multiple features simultaneously (110; 111). Dysarthric speech may result in interpersonal communication problems; yet, higher-functioning children can express themselves fully and may participate in their treatment. The language pattern is also characteristic and consists of repeated ambivalent statements that are most commonly accompanied by anxiety and coprolalia. Because intelligence test scores are influenced by difficulty in testing the subjects due to their movement disorder and dysarthric speech, overall intelligence may be underestimated in patients with Lesch-Nyhan disease (09).
Testing of classic and variant cases of Lesch-Nyhan disease revealed qualitatively similar cognitive-deficit profiles in both patient groups, but the variants produced test scores that were intermediate between those with classic Lesch-Nyhan disease and normal participants (148).
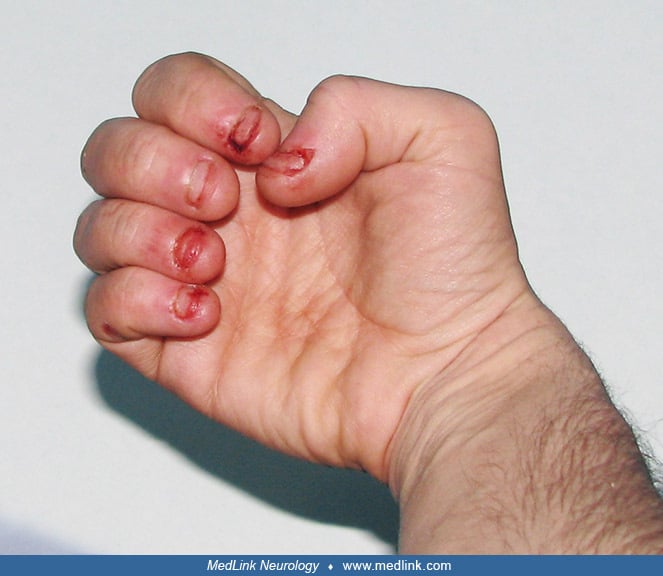
Self-injury may begin as early as 1 year of age and occasionally as late as the teens (178; 108). Self-injury occurs even though all sensory modalities, including pain sensation, are intact. Self-injurious behaviors in Lesch-Nyhan disease are typically sudden and violent in onset instead of gradually emerging over time as in other developmental disabilities (73).
Usually, self-injurious behavior is expressed as self-biting; however, other patterns of self-injurious behavior may emerge with time.
(Source: Ambarsari CG, Cahyadi D, Sari L, et al. Late diagnosis of Lesch-Nyhan disease complicated with end-stage renal disease and tophi burst: a case report. Ren Fail 2020;42(1):113-21. Creative Commons Attribution License. h...
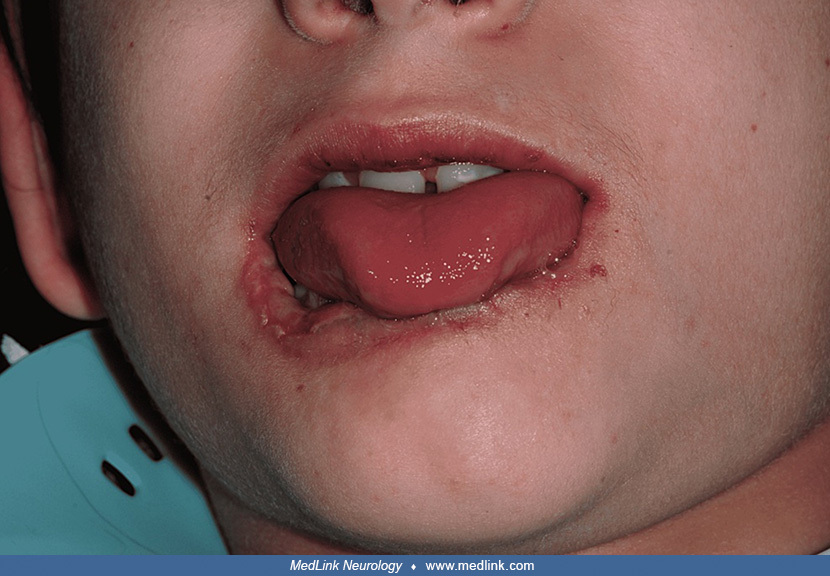
Clinical presentation of the patient with Lesch-Nyhan disease at eight months of age. An ulcer involves the tip of the tongue with well-defined regular borders and a yellowish central membrane. The lower lip exhibits deformity....
A deformed lower lip with an extensive ulcer involving both the oral mucosa and vermillion lip border in a patient with Lesch-Nyhan disease. The ulcer has well-defined regular borders and a central yellowish membrane. A white h...
(Source: Eita AA. Congenital anomalies-associated Riga-Fede disease as an early manifestation of Lesch-Nyhan syndrome: rare entities in the same pediatric patient-a case report. BMC Oral Health 2022;22(1):26. Creative Commons A...
Lip manipulation (sucking/biting) and saliva over the perioral tissues are evident. (Source: Eita AA. Congenital anomalies-associated Riga-Fede disease as an early manifestation of Lesch-Nyhan syndrome: rare entities in the sam...
When 64 families were surveyed regarding self-injury of their family members, the most common initial mode of self-mutilation and the most frequently cited past or current behavior was biting of lips or fingers (142), whereas in other intellectual disability syndromes of self-injury, self-hitting and head banging are the most common initial presentations.
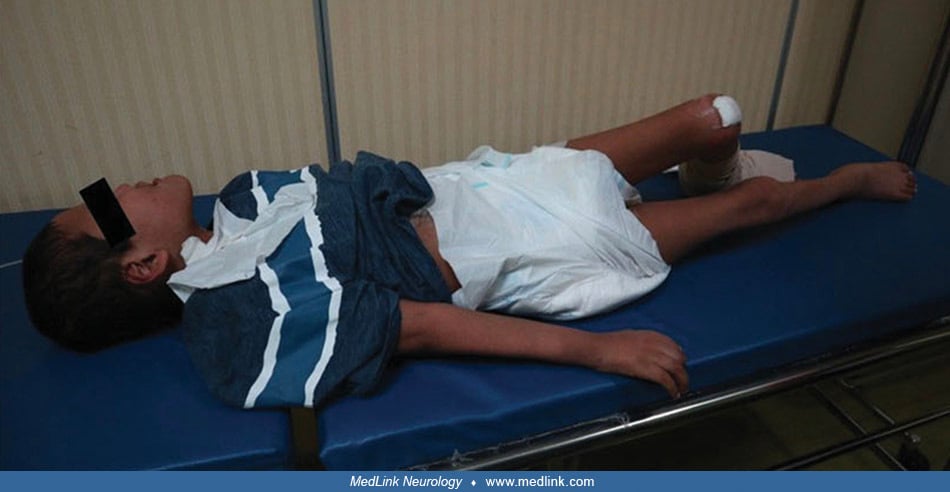
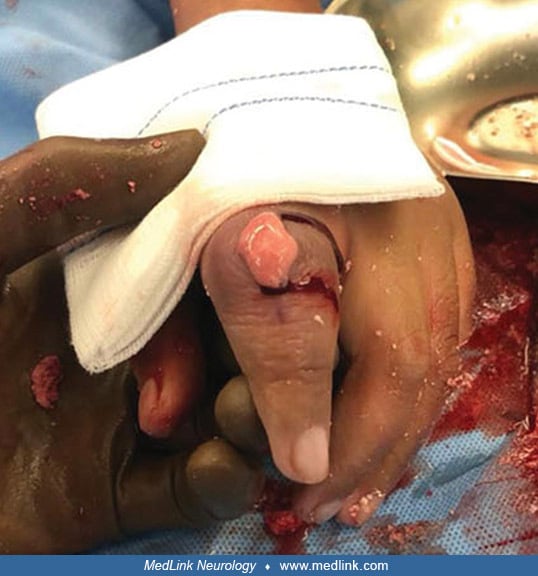
Characteristically, the fingers, mouth, and buccal mucosa are mutilated (178; 18; 108). Self-biting is intense and causes tissue damage, often leading to amputation of fingers and loss of tissue around the lips. The biting pattern is often asymmetrical in that the patient preferentially mutilates the left or right side of the body. Moreover, the intensity of the self-injurious behavior generally requires that the patient be restrained, and the biting of the lips may lead to the extraction of primary teeth as an act of desperation in the hopes of limiting further self-injury (107).
Despite the movement disorder, when restraints are taken away, the individual with Lesch-Nyhan disease may appear terrified and may quickly and accurately place a finger in the mouth. The patient may ask for restraints to prevent elbow movement and self-injury, and when the restraints are placed or replaced, the patient may appear relaxed and more good-humored (119).
Self-mutilation has been conceptualized as a compulsive behavior that the child tries to control but generally is unable to resist; this does not explain why this specific compulsion routinely develops in this disorder. In older children, self-injury may progress to deliberate self-harm and compulsive aggression toward others. The individual with Lesch-Nyhan disease may inflict injury on others through pinching, grabbing, or hitting or by using verbal forms of aggression. Afterward, he may apologize for this behavior and say that it was out of his control. Older individuals become more adept at finding ways to control self-injury, including enlisting the help of others and notifying them when restraints can be removed. Other associated maladaptive behaviors may develop later, including head or limb banging, eye poking, pulling of fingernails, and psychogenic vomiting.
Among 22 patients with varying degrees of HPRT deficiency, patients with severe enzyme deficiency had grossly abnormal ocular motility, whereas patients with moderate enzyme deficiency had clinically normal ocular motility (90). In patients with classic Lesch-Nyhan disease (ie, HPRT less than 1%), target fixation was interrupted by frequent saccadic intrusions toward minor visual distractions. Voluntary saccades were associated with an initial head movement or eye blink in all of these patients. When head motion was prevented, voluntary saccades were frequently delayed and, at times, absent. In contrast, saccade velocity, reflexive saccades, and other reflexive eye movements appeared normal. Such ocular motility disturbances are consistent with dysfunction of the basal ganglia or its connections with ocular motor centers in the prefrontal cortex or midbrain.
Lesch-Nyhan variants. Self-injury is the characteristic feature of classic Lesch-Nyhan disease, but it is not characteristic of Lesch-Nyhan variants (ie, with HPRT enzyme activity of more than 2%). In a study of 22 patients with Lesch-Nyhan disease, 11 with Lesch-Nyhan variants, and 11 healthy controls, those with a diagnosis of Lesch-Nyhan disease had severe difficulties with self-injury, aggression toward others, anxious-depressed symptoms, distractibility, motor stereotypies, and disturbing interpersonal behaviors; those with Lesch-Nyhan variants did not show self-injury and were intermediate between Lesch-Nyhan disease cases and controls on several scales (151).
A prospective multicenter international study of Lesch-Nyhan disease variants examined neurologic manifestations in 46 patients 3 to 65 years of age from 34 families (86). Motor abnormalities were found in 42 patients (91%) and ranged from subtle clumsiness to severe and disabling generalized dystonia. Cognitive functioning was affected in 31 patients (67%) but was never severe. None of the patients showed self-injurious behaviors; however, maladaptive behaviors were common. Only three patients showed no evidence of neurologic dysfunction. They were most similar to classic Lesch-Nyhan disease cases in regard to their attention deficits.
Gout is more frequent in attenuated variants than in Lesch-Nyhan disease (108; 117). Gout in attenuated variants may be associated with hyperuricemia, renal microcalculi or calculi, renal failure, and a family history of juvenile gout (117).
Variants may present with renal failure due to uric acid lithiasis and nephropathy and may be responsible for previously unexplained renal disease in multigeneration kindreds (12; 186).
Understanding the molecular disorder has led to effective drug treatment for those aspects of the disease related to uric acid accumulation and subsequent arthritic tophi, renal stones, and neuropathy. However, the overall prognosis of Lesch-Nyhan disease is poor; patients rarely survive into their third decade, with mortality typically resulting from respiratory infections, renal failure, or sudden unexpected death (116). Reduction in uric acid has not influenced the neurologic and behavioral aspects of Lesch-Nyhan disease (116); children treated from birth for uric acid elevation have behavioral and neurologic symptoms, despite never having high uric acid levels. The most significant complications are renal failure and self-mutilation.
A Caucasian male with classic Lesch-Nyhan disease presented with mood lability, a dystonic movement disorder, self-injurious behavior, and deliberate self-harm. He was first diagnosed at 2.5 years of age.
There was a family history of Lesch-Nyhan disease in six of the patient’s male family members, including his maternal great-uncle, two maternal uncles, two maternal cousins, and his older brother. The patient’s brother died of pneumonia at 18 years of age. The patient’s mother, grandmother, and maternal aunt were HPRT1 mutation carriers, as was the 16-year-old first cousin. There was no family history of depression or suicidal ideation.
There was concern with the pregnancy because the mother had two brothers with a then-unknown neurologic disease, and her first child was affected; at the time of the patient’s birth in 1964, the syndrome had not yet been reported in the medical literature.
The patient was the product of a normal, full-term pregnancy and delivery; birth weight was 8 pounds. He returned home with his mother at age 3 days. His mother became concerned when he was 3 to 4 months old because he was floppy and could not hold things in his hands. Because of his floppiness, a towel was wrapped around his waist to hold him in a highchair. At age 6 to 7 months, he began to hold onto objects. His mother ground his food at home because he could not feed properly. He began having tonic-clonic seizures at age 3 to 4 months and was started on phenobarbital (his older brother also had a seizure disorder). As he grew older, he began to crawl and speak, but he did not speak well enough to be understood until 6 years of age.
Following an ultimatum from the patient’s father that the children leave the home or he would, the patient was placed, undiagnosed, at a state hospital at age 2 years along with his older brother, who was then 3.5 years old.
Around 2.5 years of age, he was seen by American rheumatologist and biochemical geneticist Jay Seegmiller from the National Institutes of Health, who diagnosed Lesch-Nyhan disease. Seegmiller is best known for his role in discovering the biochemical basis of Lesch-Nyhan syndrome in 1967, establishing it as a disease with a defined cause (153). According to Seegmiller (personal communication to James C Harris), the patient and his brother were the fourth and fifth cases diagnosed with this disease.
The boy entered a rehabilitation program and eventually was placed in leg braces. He began to injure himself with finger biting at approximately 6 years of age. This problem persisted and required the continuous use of arm restraints. He banged his head, requiring the use of a helmet to prevent injury. However, he had not bitten his fingers for 2 or 3 years and never bit his lips, so his teeth were not removed as is the case with many other affected individuals.
He remained at the center until his adulthood with regular visits from his mother, and at 22 years of age, he graduated from high school. As an adult, he entered an alternative living program that allowed him to remain in the community. He lived in an apartment with community support and attended a day program that included computer training; he made greeting cards and banners.
As an adult, he had multiple psychiatric admissions and had been seen for outpatient counseling for mood and behavior problems linked to his demoralization and depression. His caregiver reported, “He goes through depressed times and wants to harm himself about every 6 months.” He was periodically demoralized and said he was tired of being in “that body.” When frustrated or annoyed, he harmed himself by throwing himself from his wheelchair. He attempted suicide by trying to jump out of a car, and on another occasion, he got a knife and threatened to kill himself. He was aware of the impulse to injure himself and tried to avoid it or to elicit the help of others in doing so, but he warned them not to get too close to him. He could hurt others with head thrusts or head butting, and at times he cursed at others and used inappropriate sexual talk. If he did harm or upset others, he showed remorse and apologized.
Lesch-Nyhan disease is an inborn error of metabolism with multisystemic involvement, including neurologic, skeletal, urinary, and skin components. Mutations in the HPRT1 gene, which encodes the purine salvage enzyme hypoxanthine-guanine phosphoribosyltransferase (HPRT), cause Lesch-Nyhan disease and more mildly affected Lesch-Nyhan disease variants (153; 143). HPRT enzyme is normally present in each cell in the body, but its highest concentration is in the brain, particularly the basal ganglia. HPRT is important for recycling both guanine and hypoxanthine (50). Absence of HPRT prevents the normal metabolism of hypoxanthine, resulting in excessive uric acid production and manifestations of gout and renal dysfunction unless specific drug treatment is carried out (ie, allopurinol).
Due to decreased xanthine oxidase activity in the brain, there are excessive levels of hypoxanthine and other oxypurines in the CSF; indeed, hypoxanthine levels in the CSF in individuals with Lesch-Nyhan disease are four times higher than controls, and even higher than serum levels in the same patient (116). High hypoxanthine levels may contribute to the neurologic deficits associated with Lesch-Nyhan disease (116); in cell culture studies, cells treated with excess hypoxanthine exhibited changes in essential dopaminergic developmental proteins.
Alterations in brain volume and neural connectivity. Individuals with classic Lesch-Nyhan disease exhibit significant reductions in both white matter (26%) and gray matter (17%) compared to healthy controls, especially in the medial inferior and frontal white matter regions (116). Atrophy is most evident in the basal ganglia, frontotemporal, and limbic regions, with relative sparing of the parieto-occipital regions. In classic Lesch-Nyhan disease, atrophy develops particularly in the ventral striatum, prefrontal areas, and temporal lobe, whereas variant Lesch-Nyhan atrophy develops in the lingual gyrus and precuneus, with sparing of frontotemporal regions.
Genetics. Because the gene responsible for Lesch-Nyhan disease is on the X-chromosome, the disease almost always occurs in males (124; 11; 29; 140; 72; 102), although there are very rare circumstances in which cases can occur in females (03).
Note that males in multiple generations are affected, but obligate female carriers are unaffected. (Source: Laróvere LE, Fairbanks LD, Jinnah HA, et al. Lesch-Nyhan disease and its variants: phenotypic and mutation spectrum of ...
Most families have a unique mutation; 302 different mutations were identified among 271 families (87; 88). Two cases with a normal HPRT1 cDNA were attributed to a defect in the regulation of the gene or a defect in HPRT1 gene expression regulation (28; 55). Although rare, in one family, a single mutation led to three different phenotypes in five related family members (80).
A review of more than 600 known genetic mutations, their influence on the gene product, and their relationship to the clinical phenotype found that the disease phenotype depends heavily on how mutations influence enzyme activity (49). However, additional genetic and nongenetic factors also influence genotype-phenotype correlations (19).
In a study comparing the "transcriptome" (ie, the set of all RNA transcripts, including coding and noncoding transcripts, in an individual or a population of cells) from human Lesch-Nyhan disease fibroblasts and normal human fibroblasts (using a microarray with 60,000 probes corresponding to the entire human genome), clusters of genes were identified that most significantly affected biological processes in Lesch-Nyhan disease cells; these affected genes were components of specific cellular processes involving the cell cycle (eg, cell division) and cellular metabolism (eg, purine catabolism and nucleic acids) (27).
MicroRNAs (miRNAs), critical in the transcriptional regulation of genes, are dysregulated in Lesch-Nyhan disease (70; 69; 71; 68; 67). HPRT1 mRNA transcripts may act as competitive endogenous RNAs (ceRNAs) engaged in multiregulatory crosstalk between key neural transcripts and miRNAs, which may explain how a gene such as HPRT1, responsible for housekeeping metabolic functions, can have pleiotropic effects on disparate genes and signal transduction pathways. (67). HPRT deficiency causes dysregulated expression of key genes essential for striatal patterning, most notably the striatally-enriched transcription factor B-cell leukemia 11b (Bcl11b) (68).
Females may be carriers of the disease gene but typically do not exhibit any symptoms (137), except in rare circumstances (03).
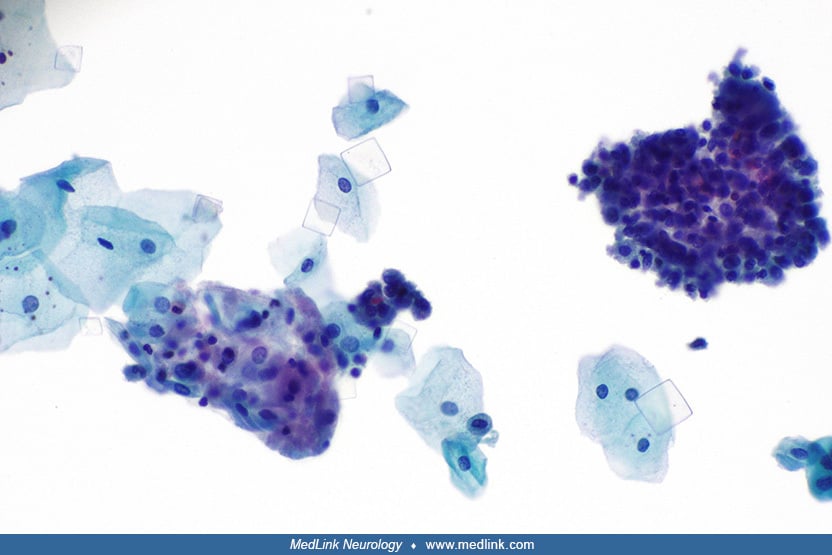
Analysis of X-inactivation of blood DNA from the affected 4-year-old girl and her parents as signified by the human androgen receptor (AR) locus.
The methylation status of the methylation-sensitive enzyme’s restriction s...
Seventy-five percent of carrier females have skewed X chromosome inactivation (169), but there have been only 14 cases of Lesch-Nyhan disease reported among females (81; 74; 124; 172; 190; 11; 185; 87; 82; 30; 29; 140; 152; 21; 03).
A wide range of genetic mutations has been detected across the female cases.
Frequency of therapeutic approaches to self-injurious oral trauma in 19 Italian patients with Lesch-Nyhan disease according to a structured telephone interview of their parents.
Key: (1) Tooth extraction. (2) Painkilling...
Among the 10 cases for whom the responsible mutation was identified, two exhibited nonsense mutations (p.Arg170* and p.Tyr153*) (11; 30); two had missense mutations (p.Glu14Lys and p.Tyr72Cys) (82; 152); two had a splice site mutation (c.609+4A> G) (87; 29; 21); one had a stop mutation (185); one carried a severe translocation (46,XX,t[X: 2][q26:p25]) (140); one had a microdeletion of HPRT1 (124); and one had a frameshift mutation (03). One case was reported twice (87; 29).
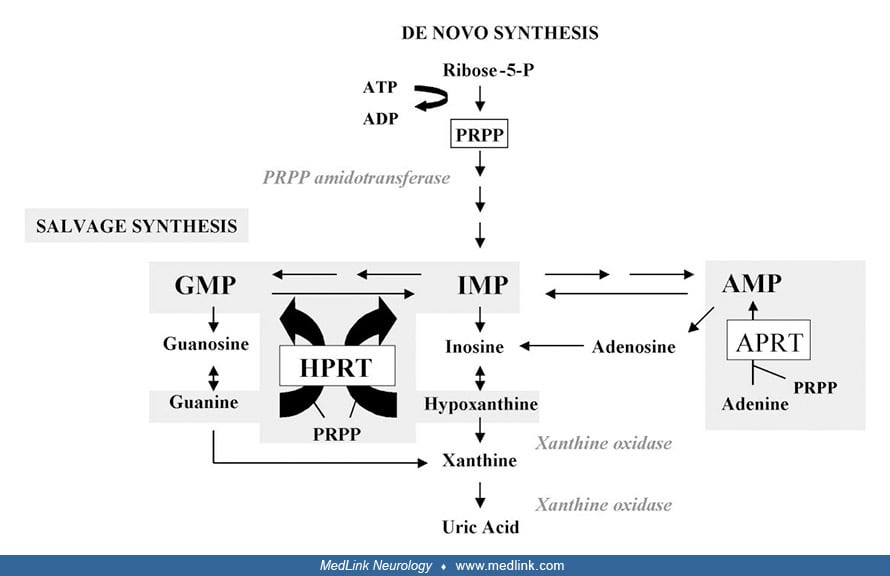
Purine metabolism. The first and rate-limiting step of de novo purine synthesis is mediated by the enzyme 5'-phosphoribosyl-1-pyrophosphate (PRPP) amidotransferase. De novo purine synthesis occurs through a multistep process and requires the contribution of four amino acids, one PRPP, two folates, and three adenosine triphosphate (ATP) molecules to synthesize an inosine monophosphate molecule.
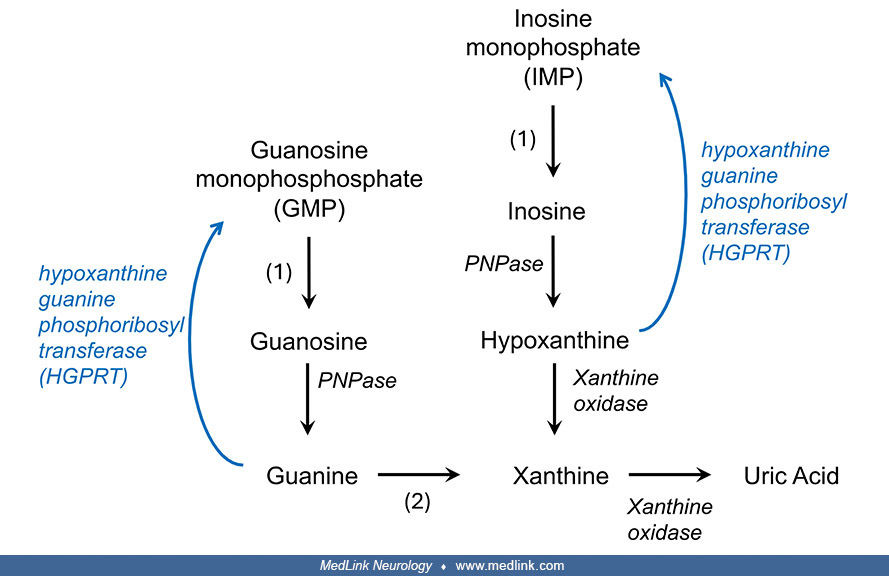
The purine salvage pathway is mediated by two enzymes: hypoxanthine phosphorybosyltransferase (HPRT) [also called hypoxanthine-guanine phosphoribosyltransferase (HGPRT)] and adenine phosphorybosyltransferase (APRT) (116). HPRT catalyzes the salvage synthesis of inosine monophosphate and guanosine monophosphate from the purine bases hypoxanthine and guanine, respectively, utilizing PRPP as a co-substrate.
HPRT deficiency results in (1) decreased purine recycling, which leads to increased purine degradation, resulting in peripheral hyperuricemia; and (2) increased activity of the de novo synthesis pathway to compensate for the loss of salvage (116). In particular, the HPRT defect in Lesch-Nyhan disease results in the accumulation of its substrates, hypoxanthine and guanine, which are converted into uric acid by means of xanthine oxidase. Xanthine dehydrogenase (EC 1.17.1.4) catalyzes the following chemical reaction:
xanthine + NAD+ + H2O <—> urate + NADH + H+
Patients with Lesch-Nyhan disease have low HGPT activity (less than 1% in striatal tissue and 1% to 2% of that of controls in thalamus cortex) but normal adenosine phosphoribosyltransferase (APRT) activity (APRT is the phosphoribosyltransferase for adenine, another amino purine), demonstrating that there is no general deficit in purine metabolism (103).
Neurotransmitter changes. Neurochemical alterations in Lesch-Nyhan disease have been assessed in multiple ways, including the following (75): (1) direct measurement of neurotransmitters in brain tissue (103); (2) measurement of neurotransmitters and their metabolites in cerebrospinal fluid (161); (3) measurement of adenosine, dopamine, and serotonin receptors in peripheral lymphocytes (54); (4) clinical response to neurotransmitter precursor treatment; and (5) measurement of the presynaptic dopamine transporter using PET scanning (184) or 18F fluorodopa (45).
Low dopamine levels and decreased levels of homovanillic acid, its main metabolite, as well as low levels of the synthesizing enzymes, dopamine decarboxylase, esterase, and hydroxylase, are found in terminal-rich dopamine areas, including the caudate nucleus, putamen, and nucleus accumbens in patients with Lesch-Nyhan disease (103; 146; 101; 20; 116). Although levodopa treatment has proven ineffective and, in some cases, worsened both motor and affective symptoms of Lesch-Nyhan disease, dopaminergic dysfunction in the basal ganglia may nevertheless contribute to the expression of dystonia and obsessive-compulsive behaviors in Lesch-Nyhan disease (103; 45; 184; 77; 116). Dopaminergic neurons of the substantia nigra pars compacta haver high purine demands and are, therefore, particularly vulnerable to metabolic disturbances, further contributing to motor dysfunction and cognitive impairment (116). Despite a functional loss of 65% to 90% of the nigrostriatal and mesolimbic dopamine terminals, the cells of origin in the substantia nigra have normal dopamine levels (103). The loss of HPRT-mediated purine recycling is associated with significant loss of dopamine and its metabolites, indicating an important connection between purine and dopamine pathways (63). An important functional link between purine nucleotides and the dopamine system involves guanine, the precursor of guanine triphosphate (155): (1) The absence of HGPT may affect dopamine systems due to limited recycling of guanine; for example, guanine nucleotide depletion induces differentiation and aberrant neurite outgrowth in human dopaminergic neuroblastoma units (115); and (2) The binding of dopamine to its receptor leads either to activation (D1 receptor) or inhibition (D2 receptor) of adenylcyclase, with both processes mediated by guanine triphosphate-binding proteins. In a genetic mouse model of Lesch-Nyhan disease, HPRT deficiency disrupts dopaminergic circuit development (182).
Other neurotransmitters are also affected in Lesch-Nyhan disease. HPRT deficiency results in elevated extracellular levels of hypoxanthine, which can bind to the benzodiazepine agonist recognition site on the GABA(A) receptor complex (36). In addition, guanine-based purines may be important regulators of the synaptic availability of L-glutamate (36).
Thus, both neurodevelopmental (eg, a failure to "arborize" dopaminergic synaptic terminals) and neurotransmitter (eg, disruption of GABA and glutamate receptor-mediated neurotransmission) consequences of Lesch-Nyhan disease may contribute to the disorder (103; 14; 36).
Biochemical-clinical relationships. Classification of cases across the clinical spectrum of HPRT activity has been attempted based on neurologic symptoms, dependency for personal care, HPRT activity in hemolysate and in intact erythrocytes, and predicted protein size (138):
|
• Group 1: normal development with no neurologic symptoms, HPRT activity detectable in both hemolysates and intact erythrocytes, and the mutation did not affect the predicted protein size. | |
|
• Group 2: mild neurologic symptoms that did not prevent independent living, HPRT activity detectable in intact erythrocytes, and normal protein size. | |
|
• Group 3: severe neurologic impairment that precluded independent living, no residual HPRT activity, and normal protein size. | |
|
• Group 4: clinical characteristics of Lesch-Nyhan syndrome (although some may not show self-injurious behavior), no residual HPRT activity, and typically (although not universally) an altered predicted protein size. |
There are widespread cerebral changes in Lesch-Nyhan disease. A study utilizing volumetric MRI in seven patients with Lesch-Nyhan disease demonstrated a 34% decrease in caudate volume and a 17% decrease in total cerebral volume compared with normal controls (78). Another study involving 21 classic cases, 17 variant cases, and 33 control subjects documented neuroanatomical abnormalities that extend beyond the basal ganglia (149); compared to healthy controls, in patients with classic Lesch-Nyhan disease and variant cases that involve the basal ganglia, limbic, and frontotemporal regions, there was sparing of the parieto-occipital regions. In addition, there are substantial white matter volume abnormalities in Lesch-Nyhan disease that may be more severe than gray matter deficits in classic Lesch-Nyhan disease and also characterize variant cases (150; 31).
Self-injurious behavior in Lesch-Nyhan disease is not caused by hyperuricemia or excess hypoxanthine; patients with partial variants with hyperuricemia do not self-injure (02), and infants with Lesch-Nyhan disease who were treated for hyperuricemia alone from birth still develop self-injurious behavior. Uric acid is not produced in the brain and does not cross the blood-brain barrier (104). Thus, uric acid is not responsible for brain dysfunction; purine nucleotide depletion or accumulation of other toxic purine intermediates could be more relevant (104). Self-mutilation in Lesch-Nyhan disease is caused by dopaminergic denervation (61).
Attenuated clinical phenotypes are associated with residual HPRT activity, whereas the most severe phenotype is usually associated with null activity (130; 132). In cases of gouty arthritis with urate overproduction, a careful evaluation for motor impairments or neurocognitive abnormalities may help to identify attenuated variants of Lesch-Nyhan disease (49; 117).
Hypoxanthine and guanine recycling and the resultant compensatory changes in the de novo synthesis of purines are important to understanding the full pathogenesis of Lesch-Nyhan disease (51). Loss of guanine recycling may be more tightly linked to Lesch-Nyhan disease or variant phenotype than loss of hypoxanthine recycling (154; 147). Moreover, patients with inherited GTP cyclohydrolase deficiency show clinical features of dystonia in common with Lesch-Nyhan disease (170; 53; 37; 52).
HPRT activity is related to the extent of motor symptoms, presence or absence of self-injury, and possibly to the degree of cognitive function (132). Partial deficiency in HPRT (Kelley-Seegmiller disease) is associated with hyperuricemia and variable neurologic dysfunction (109; 138). Most commonly, partial HPRT deficiency leads to a severe form of gout. However, in one study, five of nine partial variant patients (HPRT levels 1.8 to 20%) had IQ scores below 70 and had deficits in working memory (148). Due to the enzyme deficiency, hypoxanthine accumulates in the cerebrospinal fluid, but uric acid does not.
Early histopathologic and electron microscopic studies found nonspecific abnormalities in Lesch-Nyhan disease, including loss of neurons and gliosis of the cerebral cortex and focal demyelination of the cerebral white matter (178). A subsequent study of neuropathological findings in 33 cases (two new cases and 31 previously reported cases) found that a small cerebrum (39%) and chronic cerebellar lesions (27%) were the most frequent abnormalities (32).
Histopathological studies using autopsy tissue from five Lesch-Nyhan disease cases and six controls found no signs suggestive of a degenerative process or other consistent abnormalities in any brain region, although neurons of the substantia nigra in the cases showed reduced melanization and reduced immunoreactivity for tyrosine hydroxylase, the rate-limiting enzyme in dopamine synthesis (64). A comparison HPRT-deficient knockout mouse model showed reduced tyrosine hydroxylase expression in the striatum and flow-activated cell sorting in a cell model of HPRT deficiency, revealed significantly reduced tyrosine hydroxylase immunoreactivity compared to the control parent line. These results are consistent with a neurochemical phenotype involving dopaminergic neurons not linked with a degenerative process.
Selected findings in animal models. HPRT deficiency is strongly associated with a loss of basal ganglia dopamine (92; 89). During the first 8 weeks of postnatal development, dopamine levels in whole-brain extracts from the mutant HPRT-deficient mice failed to increase at rates comparable to normal animals resulting in 40% lower dopamine levels throughout adulthood (92). Regional analysis in adult animals showed dopamine levels to be most severely depressed in the caudoputamen and less severely and less consistently depressed in the mesolimbic area, whereas other neurotransmitter systems appeared relatively unaffected (92; 89). These results suggest an abnormality in the dopamine system despite apparently normal spontaneous behavior.
Both HPRT-deficient mice and "doubly-deficient" HPRT-APRT-deficient mice devoid of any purine salvage pathways exhibit none of the behavioral symptoms associated with the human Lesch-Nyhan syndrome, including self-injurious behavior (44).
A relationship between dopaminergic supersensitivity and self-injury has been demonstrated in an experimental animal model (16). When neonatal (5-day-old) and adult rats that were not HPRT-deficient were administered 6-hydroxydopamine to denervate basal ganglia regions, the age when neural function is disrupted determined the type of motor and behavioral symptoms observed. Neonatally treated rats exhibited self-biting and self-mutilation when challenged as adults with either L-dopa or a D1-receptor agonist, whereas this self-injurious behavior was not seen in adult rats treated with 6-hydroxydopamine. Self-biting in neonatally exposed rats was blocked by a D1 dopamine antagonist. Repeated stimulation of D1 receptors in adulthood ("D1 priming") interacts with a developmental loss of dopamine to profoundly and persistently modify neuronal signaling and dendrite morphology in the mature prefrontal cortex in the absence of associated markers of neurodegenerative change (135).
|
• A population study in the United Kingdom estimated the prevalence of classic Lesch-Nyhan disease as 1 in 2 million. | |
|
• Over 20 years of study, the mean incidence rate in the United Kingdom was 0.18 per 100,000 live births. | |
|
• Those with the classical form of the disease rarely survive the third decade; however, lifespan may be normal for those with partial variants without severe renal involvement. |
Early estimates of the prevalence of classic Lesch-Nyhan disease were 1 in 100,000 to 1 in 380,000 (25). However, based on the number of known cases in the United States, it may be as rare as 1 in 800,000 to 1 in 1.2 million. A population study in the United Kingdom estimated the prevalence of classic Lesch-Nyhan disease as 1 in 2 million (112).
Over 20 years of study, the mean incidence rate in the United Kingdom was 0.18 per 100,000 live births (112). The incidence of partial variants is not known.
Those with the classical form of the disease rarely survive the third decade; however, lifespan may be normal for those with partial variants without severe renal involvement.
|
• Precise molecular genetic methodologies are available for carrier and prenatal diagnosis. | |
|
• Prenatal diagnosis is available for mothers of affected children by measurement of HPRT and APRT in chorionic villus samples or in cultured amniotic fluid cells. | |
|
• Preimplantation diagnosis with in vitro fertilization has been successfully accomplished. |
Precise molecular genetic methodologies are available for carrier and prenatal diagnosis (04). Due to the large number of known mutations, a cloning assay has been developed that allows determination of carrier status when the HPRT1 mutation is not known or is difficult to determine; carrier status can be determined in 10 days (129). As this condition is extremely rare, this approach is used primarily in those families with an affected family member.
Because there may be delays in determining the mutation, there continues to be a place for enzyme analysis (123). Although uncultured chorionic villi may be used for prenatal diagnosis by direct enzyme assay, the use of cultured amniocytes or chorionic villi cells for enzyme assay is preferable (121). For mutation carrier status, a peripheral blood T lymphocyte cloning assay uses resistance to the purine analog 6-thioguanine to measure the frequency of cells in females expressing a mutant HPRT1 allele (129). This assay allows determination of the carrier status of females even when the HPRT1 mutation is not yet known or is difficult to determine. Information on carrier status is available within 10 days.
Prenatal diagnosis is available for mothers of affected children by measurement of HPRT and APRT in chorionic villus samples or in cultured amniotic fluid cells (65). Preimplantation diagnosis has been offered in some medical centers for couples who prefer selection prior to pregnancy rather than during pregnancy (15; 33). Preimplantation diagnosis with in vitro fertilization has been successfully accomplished (139; 24).
Germline mosaicism may rarely complicate molecular diagnosis of Lesch-Nyhan syndrome (179).
The diagnosis of Lesch-Nyhan disease is straightforward when fully developed with the classic clinical triad: uric acid overproduction, neurologic dysfunction, and cognitive and behavioral disturbances (85). There are no other genetic causes of the full phenotype. The main diagnostic difficulties arise in two situations: (1) during the early stages when all features are not yet apparent; and (2) in individuals who have milder phenotypes (85). The index of suspicion for HPRT deficiency should be raised when developmental delay is associated with evidence of overproduction of uric acid, such as hyperuricemia, uric acid nephrolithiasis, urate crystals in the urine, or gouty arthritis (85).
The differential diagnosis includes other causes of infantile hypotonia and dystonia. Most commonly, children are misdiagnosed as having athetoid cerebral palsy due to similar age of onset and associated movement disorders (eg, athetosis and dystonia) (116). When a diagnosis of cerebral palsy is suspected in an infant with a normal prenatal, perinatal, and postnatal course, Lesch-Nyhan disease should be considered.
Lesch-Nyhan disease is often first suspected when self-injurious behavior emerges. However, self-injurious behaviors occur in other conditions, including psychiatric diseases (eg, schizophrenia, obsessive-compulsive disorder) and autism spectrum disorder.
Self-injurious behavior can also be seen in other neurodevelopmental, neurometabolic, and neurodegenerative disorders as well as in disorders with characteristic structural brain changes and heterogeneous etiologies (eg, autoimmune and drug-induced) (48; 85). These include nonspecific intellectual disability, Tourette syndrome, McLeod neuroacanthocytosis syndrome (chorea-acanthocytosis), Rett syndrome, Cornelia de Lange syndrome, glutaric acidemia type 1, familial dysautonomia, and sensory neuropathy (eg, hereditary sensory neuropathy type 1) (85).
Finger and lip biting is so severe and so characteristic of Lesch-Nyhan disease that it is referred to as a behavioral phenotype (85). In other conditions associated with self-injury, the behavior tends to be less severe (85). In addition, in other conditions with self-injury behaviors, the range of behaviors is different, usually with head banging or nonspecific self-biting--but not biting of the fingers and lips to a degree that results in tissue damage as is the case with Lesch-Nyhan disease (85).
|
• The preliminary diagnosis is ordinarily based on the physical and behavioral phenotype. | |
|
• The presence of dystonia in conjunction with self-injury and hyperuricemia is usually diagnostic. | |
|
• Serum levels of uric acid that exceed 4 to 5 mg uric acid/dL and a urate-to-creatinine ratio of 3 or more are highly suggestive of HPRT deficiency, especially when associated with neurologic symptoms. | |
|
• Definitive diagnosis requires an analysis of HPRT enzyme activity. | |
|
• Molecular genetic techniques are used primarily for the identification of carriers. |
Early diagnostic genetic testing can play an important role in helping families make difficult decisions (192). Given the implications of Lesch-Nyhan disease diagnosis, the management complexity of the disease, and the limitations of available therapies, parental values must guide clinical decision-making (192). Careful and empathic discussion of such rare genetic diagnoses with families is complex but essential for facilitating critical discussions to empower families to make informed decisions (192).
Laboratory studies. The preliminary diagnosis is ordinarily based on the physical and behavioral phenotype. Orange crystals present in a child’s diaper are typically the first indications of Lesch-Nyhan disease (116). The presence of dystonia in conjunction with self-injury and hyperuricemia is usually diagnostic. With partial deficiency, the diagnosis will be less clear. In virtually all cases, hyperuricemia and increased secretion of uric acid are present even at birth (116). Serum levels of uric acid that exceed 4 to 5 mg uric acid/dL and a urate-to-creatinine ratio of 3 or more highly suggest HPRT deficiency, especially when associated with neurologic symptoms. However, in contrast, there are relatively normal levels of uric acid in the CSF due to decreased xanthine oxidase activity in the brain (116).
Definitive diagnosis requires an analysis of HPRT enzyme activity in an erythrocyte lysate (85). Patients with classic Lesch-Nyhan disease have enzyme activity near 0%, whereas variant cases have values between 0% and 60% (130). The enzyme activity in partial variants shows little correlation with the clinical phenotype (155). The intact cell assay in skin fibroblasts provides a good correlation between enzyme activity and the severity of the disease (131).
Molecular genetic techniques are also used, primarily in the identification of carriers (155; 04; 85). Sequence analysis of HPRT1 is performed to detect small intragenic deletions or insertions and missense, nonsense, and splice-site variants (85). If no variant is detected by the sequencing method, the next step is to perform gene-targeted deletion and duplication analysis to detect exon and whole-gene deletions or duplications. A multigene panel that includes HPRT1 is most likely to identify the genetic cause of the condition while limiting the identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype (85). The proportion of probands with a pathogenic variant detectable by sequence analysis is approximately 80%, whereas the proportion of probands with a pathogenic variant detectable by gene-targeted deletion and duplication analysis is approximately 20% (85).
Female heterozygotes also have purine overproduction and enhanced purine nucleotide degradation (137). An elevated hypoxanthine or xanthine excretion rate differentiates most heterozygotes for HPRT deficiency from noncarrier women (137).
Macrocytic erythrocytes occur in 81% to 92% of subjects with Lesch-Nyhan disease or its neurologic variants (17). After excluding cases with iron deficiency (that may pseudonormalize erythrocyte volumes), macrocytosis occurred in 97% of subjects (17). Macrocytic erythrocytes were sometimes accompanied by mild anemia but rarely by severe anemia (17). Macrocytosis is so characteristic that its absence should prompt suspicion of a secondary process (eg, iron deficiency) in patients with Lesch-Nyhan disease (17). Because macrocytosis is uncommon in unaffected children, it may also be a clue for early diagnosis of Lesch-Nyhan disease in children with neurodevelopmental delay. Recognition of this characteristic feature of Lesch-Nyhan disease should help prevent unnecessary diagnostic testing and inappropriate treatment with folate or B12 supplements.
Recommended laboratory studies after the initial diagnosis of HPRT-deficiency disorders include: (1) serum uric acid, (2) serum chemistries including BUN and creatinine, (3) complete blood count, (4) renal function studies, and (5) urinalysis (85).
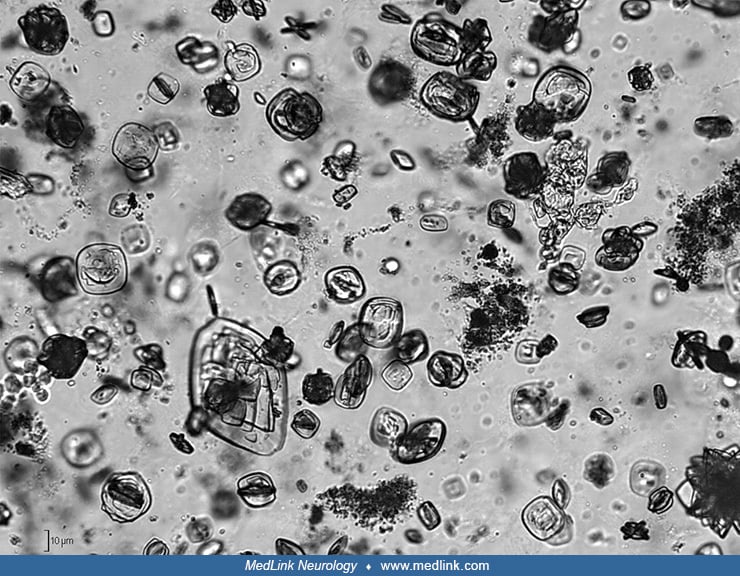
Urinary stones. Urinary stones in patients with Lesch-Nyhan disease are typically composed of hydrogen urate, but urine may contain either urate or struvite (magnesium ammonium phosphate) stones or crystals (127; 102) or xanthine stones or crystals.
Uric acid stones form in acidic urine and typically in the setting of hyperuricemia. Uric acid stones are radiolucent. Uric acid crystals are rounded parallelograms or flat, four-sided plates.
Struvite crystals and stones (also known as triple phosphate stones and "infection stones") are associated with urinary tract infections. The organisms implicated in the urinary tract infections of patients with struvite stones are overwhelmingly urea-splitting, meaning they possess an enzyme, urease, to breakdown urea, which is abundant in urine (eg, Klebsiella, Proteus, Providencia, Pseudomonas, and some species of staphylococci). Struvite stones are also often referred to as staghorn calculi, based on how they typically appear in radiologic studies. Struvite calculi are radiopaque, and the microscopic appearance of struvite crystals is often described as "coffin-lid" shaped.
Patients with Lesch-Nyhan syndrome who are treated with allopurinol may also develop multiple and bilateral renal and bladder stones by precipitation of xanthine (105; 125; 127; 79; 177; 133; 160; 166; 126; 159; 114; 156). Treatment with allopurinol inhibits the formation of uric acid and modifies renal excretion of oxypurines. Under certain circumstances (ie, decreased urinary output and pH, aggressive lowering of uric acid levels), patients can precipitate forming radiolucent xanthine stones, often in conjunction with uric lithiasis (66). Xanthine stones, both in-vitro and in-vivo, appear dense on CT and echogenic on ultrasound and lack signal on MRI (156). Xanthine urolithiasis is usually a relatively benign condition, which is easy to prevent or cure by appropriate alkalinization, forced hydration, and restriction of dietary purines, but asymptomatic or unrecognized stones may cause destruction of renal parenchyma and renal failure (160; 57; 171). Even patients with Lesch-Nyhan syndrome can have renal pelvis calculi dissolve within 10 days with urine alkalinization and hydration (126).
Radiology. Cranial MRI or CT in Lesch-Nyhan disease may show mild cerebral atrophy and small caudates (78; 77; 149; 150). Volumetric studies demonstrate significant decreases in both caudate volume and total cerebral volume (78; 149; 150).
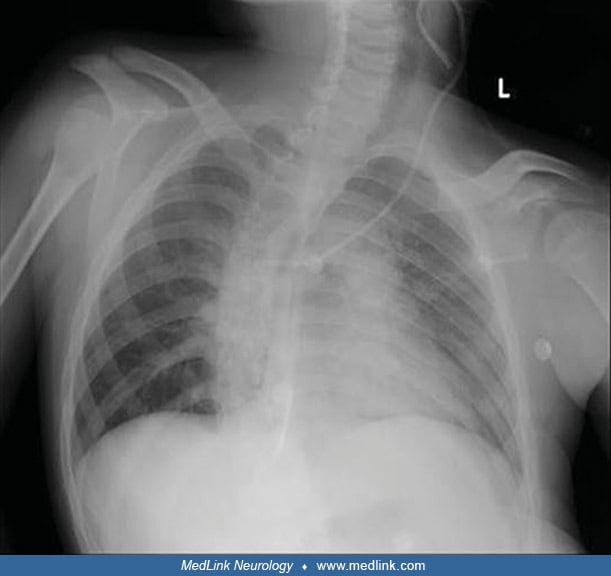
Cervical spine films may demonstrate evidence of atlantoaxial instability in Lesch-Nyhan syndrome (157).
X-rays of the limbs may show evidence of tophi in the form of soft tissue masses adjacent to joints, possibly with intramass calcifications (06).
X-ray image of the left wrist showing a soft tissue mass adjacent to the metacarpal and proximal to the second finger due to a collection of sodium urate monohydrate, or uric acid. (Source: Ambarsari CG, Cahyadi D, Sari L, et a...
The x-ray shows a large soft tissue mass with intramass calcification due to a collection of sodium urate monohydrate, or uric acid. (Source: Ambarsari CG, Cahyadi D, Sari L, et al. Late diagnosis of Lesch-Nyhan disease complic...
The x-ray shows a large soft tissue mass with intramass calcification due to a collection of sodium urate monohydrate, or uric acid. (Source: Ambarsari CG, Cahyadi D, Sari L, et al. Late diagnosis of Lesch-Nyhan disease complic...
Radiographic findings in Lesch-Nyhan syndrome commonly include faintly radiopaque stones on abdominal radiographs, and when renal disease is present, small kidneys with poor function on intravenous urogram. Radiolucent stones in patients with Lesch-Nyhan disease are usually composed of uric acid; however, several cases of xanthine and hypoxanthine-containing calculi, which may also be radiolucent, have been described in patients with Lesch-Nyhan disease receiving allopurinl therapy. Indeed, all oxypurines (ie, the collective name for the compounds of hypoxanthine, xanthine, and uric acid) may be radiolucent.
Renal parenchymal oxypurine deposition may be readily demonstrated by ultrasonography but not detected on standard radiographs or intravenous urograms (163). Therefore, abdominal ultrasound is recommended after the initial diagnosis of an HRPT-deficiency disorder to assess for renal calculi (06; 85; 156).
In some cases, abdominal CT may demonstrate stones (156).
(Source: Ambarsari CG, Cahyadi D, Sari L, et al. Late diagnosis of Lesch-Nyhan disease complicated with end-stage renal disease and tophi burst: a case report. Ren Fail 2020;42(1):113-21. Creative Commons Attribution License. h...
The stone is echogenic with posterior acoustic shadowing. (Source: Shamir SB, Peng Q, Schoenfeld AH, Drzewiecki BA, Liszewski MC. CT, US and MRI of xanthine urinary stones: in-vitro and in-vivo analyses. BMC Urol 2020;20(1):157...
The stone demonstrates posterior twinkle artifact on color Doppler imaging (right). (Source: Shamir SB, Peng Q, Schoenfeld AH, Drzewiecki BA, Liszewski MC. CT, US and MRI of xanthine urinary stones: in-vitro and in-vivo analyse...
Coronal CT image showing a stone within the left kidney along with measurement of CT number utilizing region of interest sampling. (Source: Shamir SB, Peng Q, Schoenfeld AH, Drzewiecki BA, Liszewski MC. CT, US and MRI of xanthi...
Chest radiography may show thoracic scoliosis (06).
|
• Management of Lesch-Nyhan disease incorporates psychosocial support to the patient and caregivers, behavioral and pharmacological treatment, use of protective equipment, and dental, orthopedic, and nephrology management. | |
|
• Goals of treatment in Lesch-Nyhan disease include reduction of self-mutilating and aggressive behavior, prevention of renal failure and urologic complications related to hyperuricemia, control of dystonic movements, and orthopedic correction of hip subluxation and dislocation. | |
|
• Behavioral approaches can substantially reduce self-injury and phobic anxiety associated with being unrestrained in patients with Lesch-Nyhan disease, especially if combined with stress management to assist patients in developing more effective coping mechanisms. | |
|
• The issue of restraints is complicated, even in the setting of self-injury, but parents and providers invoke this as a last resort to limit self-mutilation in Lesch-Nyhan disease. | |
|
• A nonsurgical splint or mouth guard should be the first method tried to limit self-biting in Lesch-Nyhan disease, and only if unsuccessful should more complicated devices or surgical procedures (eg, dental extraction) be considered. | |
|
• Pharmacological approaches to decrease anxiety, self-injury, and spasticity with medication have met with mixed results; there are no well-designed randomized controlled trials that support any medication for control of these problems in Lesch-Nyhan disease. | |
|
• In Lesch-Nyhan disease, the overproduction of uric acid must be controlled to prevent the development of urologic and articular complications. | |
|
• The control of uric acid in Lesch-Nyhan disease requires: (1) inhibiting the metabolism of hypoxanthine and xanthine to uric acid with allopurinol (or possibly febuxostat); and (2) generous hydration, which should be increased during periods of increased fluid loss. | |
|
• Enzyme replacement therapy, bone marrow transplant, and gene therapy are not presently viable for Lesch-Nyhan disease. | |
|
• Multiple anecdotal reports indicate improvement or resolution of self-mutilating and aggressive behavior following bilateral chronic stimulation of the globus pallidus internus for control of dystonic movements in Lesch-Nyhan disease. |
Management incorporates psychosocial support to the patient and caregivers, behavioral and pharmacological treatment, use of protective equipment, and dental management (76). Treatment goals include reduction of self-mutilating and aggressive behavior, prevention of renal failure and urologic complications related to hyperuricemia, control of dystonic movements, and orthopedic correction of hip subluxation and dislocation.
Currently, there is no standardized treatment for the neurologic manifestations of Lesch-Nyhan disease (116). A systematic review of studies assessed the efficacy of pharmacological and nonpharmacological interventions in management of neurologic symptoms in Lesch-Nyhan disease (96). Twenty-two of 34 (65%) full-text papers reviewed were rated as having a high risk of bias. Considerable heterogeneity was found in the timing of treatment implementation, adjunctive treatments, and outcome assessment. Therapeutic failures were underreported. S-adenosylmethionine and deep brain stimulation (DBS) were the most studied treatment methods and require further research to address inconsistencies. Improved and standardized study designs and reduction of publication bias are necessary to overcome current limitations of intervention efficacy.
Behavioral management. Behavioral approaches (eg, time-outs and differential reinforcement of behaviors that are not self-injurious) can substantially reduce self-injury and phobic anxiety associated with being unrestrained, especially if combined with stress management to assist patients in developing more effective coping mechanisms (39; 119; 97; 128).
Most parents identify stress reduction and awareness of the patient's needs as the most effective means of reducing self-injury (08). The most common methods used by families to minimize self-injury are attending to physical comfort, judicious use of restraints, talking to the child, distraction (eg, finding something more interesting), and attending to their specific needs (08). Such positive behavioral techniques of reinforcing appropriate behavior were rated effective by almost half of the families. The least commonly used interventions are negative behavioral approaches, such as stopping talking to the child, turning away from the child, or taking away something the child likes.
Anxiety may manifest through posturing, facial grimacing, and increases in choreoathetoid and spastic movements. The patient should be allowed to respond and give permission before proceeding with the behavioral program. Relaxation procedures may be taught, particularly those that encourage the patient to relax various body parts. Music may be utilized in conjunction with stress-reducing exercises. As with any patient, careful explanations (presented at a cognitive level that the patient can understand) should be given about what is to follow when initiating a medical or behavioral procedure.
Individuals with Lesch-Nyhan disease do not respond favorably to contingent electric shock that suppresses self-injury in other contexts. Indeed, in this disorder, an increase in self-injury is observed when aversive methods (eg, shock) are utilized (39; 07).
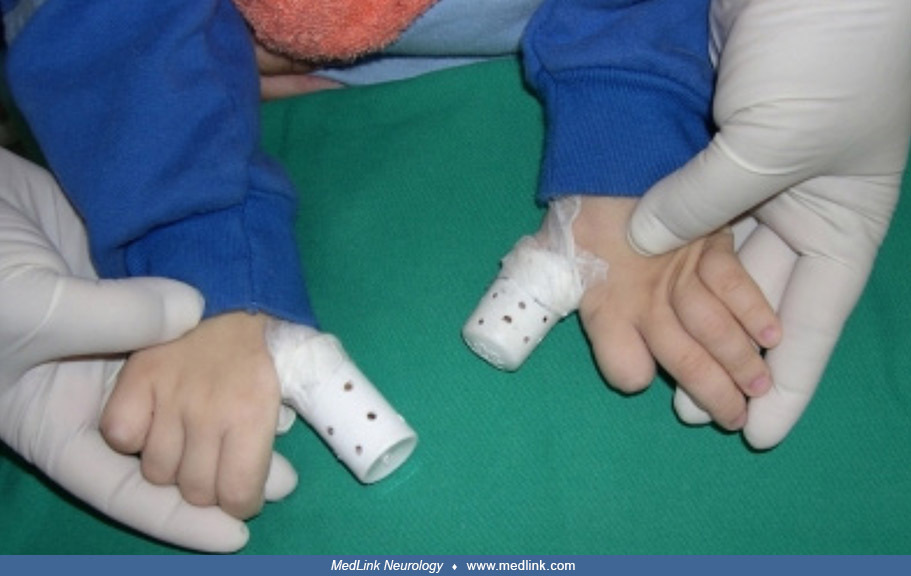
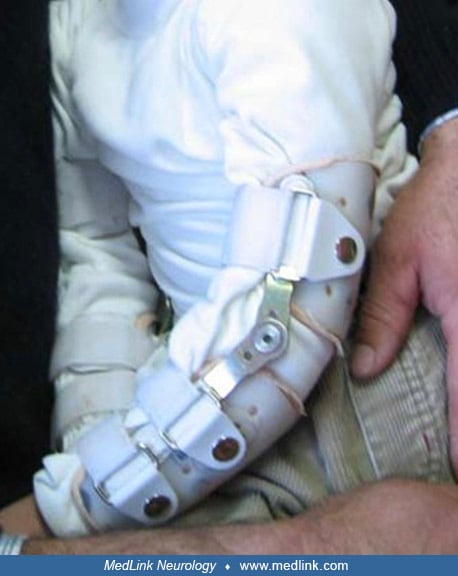
Restraints to prevent self-injury. The issue of restraints is complicated, even in the setting of self-injury, but parents and providers invoke this as a last resort to limit self-mutilation. Typically, restraints are applied to the hips, chest, elbows, hands, or specific fingers (107; 116). Wrapping the hands, applying distal orthotics, and obscuring the hands from view may all help to decrease the compulsive urge to self-harm (107; 116). In severe cases, the use of mouth guards or tooth removal may be necessary to prevent lip and tongue biting.
(Source: Ferrao J, Rodrigues Barros C, Figueiredo L, Fernandes A. Oral self-mutilation in Lesch-Nyhan syndrome: a case report. Cureus 2022;14[8]:e27874. Creative Commons Attribution License [CC BY], https://creativecommons.org/...
Elbow restraints allow hand use without the possibility of finger mutilation (168).
A survey of the families of 40 patients found that 45% of the patients were constantly restrained to limit self-injury, whereas 20% were restrained 75% of the time (08). Nearly three-fourths (72%) were always restrained at night. Only 12% were never in restraints. The older the patient was at the onset of self-injury, the less time was used for restraint. Half of the patients asked to be restrained in a particular way, and 42% insisted on techniques that a priori would be considered ineffective (eg, a lightweight glove, small bandage, or something else that could not physically prevent biting). When family members were surveyed about the patient's attitude toward restraints, 88% said the patient was at ease when restrained and liked to be restrained. Only three patients (0.7%) said they did not want to have restraints.
Botulinum toxin is useful and safe for treating self-biting behavior in patients with Lesch-Nyhan disease (59; 56), but this should be considered a pharmacologic form of restraint.
Dental management to reduce self-injury. Compulsive self-mutilation includes, in particular, biting behavior on the oral mucosa, tongue, lips, fingers, and shoulders, typically before 1 year of age. Unfortunately, no standard methods have been established to prevent self-biting in Lesch-Nyhan disease (26). In general, preventive dental treatments must be developed for each patient based on clinical findings.
Conservative approaches to preventing self-harm in patients with Lesch-Nyhan disease should be tried before radical approaches are employed (47). For example, from the age of 8 years, a boy with Lesch-Nyhan disease displayed self-harm behavior, with finger and oral injuries. He presented at age 12 with deep ulcerated lesions and tissue loss of the lips and tongue, associated with a significant decrease in food intake and consequent weight loss (47). To delay radical treatments, conservative options were initially employed (ie, therapy with gabapentin, lorazepam, and botulinum toxin injections), which successfully reduced self-mutilation, facilitated wound healing, and produced a significant improvement in nutritional status at the follow-up visit 2 months later.
In general, a nonsurgical splint or mouth guard should be one of the first approaches tried (168), and only if unsuccessful should more complicated devices (46) or surgical procedures (eg, vital pulpotomy and coronal resection or dental extraction) be considered (98).
Although dental devices are recommended, failure rates are high (08; 62). In one survey, 15 of 40 patients (38%) had previously tried a protective mouth guard designed by a dentist, but this was deemed helpful in only 20% of the selected group, whereas in contrast, 24 patients (60%) had teeth extracted to prevent self-injury, which the families generally endorsed as necessary and helpful (08). In another smaller study, among five patients with Lesch-Nyhan disease who used mouth guards to spare their teeth, dental extraction was ultimately required in four cases (62).
Most of these patients eventually require several procedures, including dental extractions, to prevent significant secondary lesions (83). In a study of 19 Italian patients, using structured telephone interviews, 71% had tooth extractions, 36% used obstructive methods (eg, placing objects between the teeth to prevent biting lesions), and 29% used orthodontic appliances (eg, an acrylic maxillary device, designed and constructed with an occlusal plate raising the bite, or a soft resin mouth guard), whereas just 14% used dental recontouring (ie, filing the teeth to make them less likely to cut the oral and perioral tissues) (83).
Frequency of therapeutic approaches to self-injurious oral trauma in 19 Italian patients with Lesch-Nyhan disease according to a structured telephone interview of their parents.
Key: (1) Tooth extraction. (2) Painkilling...
Pharmacotherapy. Pharmacological approaches to decrease anxiety, self-injury, and spasticity with medication have met with mixed results; there are no well-designed randomized controlled trials that support any medication for control of these problems in Lesch-Nyhan disease.
Drugs to control hyperuricemia. In Lesch-Nyhan disease, the overproduction of uric acid must be controlled to prevent the development of urologic and articular complications; however, reducing serum uric acid levels does not ameliorate neurologic symptoms (116). The goal is to maintain uric acid levels in the normal range without suppressing uric acid below normal levels, which risks the development of oxypurine stones instead. Oxypurine is the collective name for the compounds hypoxanthine, xanthine, and uric acid--all of which may be radiolucent.
The control of uric acid requires: (1) inhibiting the metabolism of hypoxanthine and xanthine to uric acid with a xanthine oxidase inhibitor allopurinol (or possibly febuxostat); and (2) generous hydration, which should be increased during periods of increased fluid loss (eg, febrile illness or recurrent emesis). The dosage of allopurinol, a xanthine dehydrogenase (formerly called xanthine oxidase) inhibitor, must be carefully monitored because excessive dosage can result in xanthinuria and xanthine urolithiasis (167). Excessive doses of allopurinol may result in acute renal failure from xanthine urolithiasis (160).
Drugs to control self-injury and agitation. Very little high-quality information supports pharmacological approaches to self-injurious behavior in patients with Lesch-Nyhan disease (145). In general, drug treatments have not been successful in eliminating self-injury, including the use of diazepam, haloperidol, fluphenazine, clomipramine, L-dopa, and pimozide (178). For a discussion of botulinum therapy see “Restraints to prevent self-injury” section above.
In a parent survey, 35 of 40 patients (88%) had used medication for self-injury or agitation (08). Seven classes of medications had been tried: benzodiazepines, neuroleptics, antidepressants, opiate antagonists, chloral hydrate, beta-blockers, and carbidopa-levodopa. The medications parents reported to be most effective were benzodiazepines (especially diazepam).
An open-label trial of levodopa showed no benefit, and all six participants stopped the medication early because of loss of effect or worsening of motor symptoms (176). However, in a small case series of three patients, levodopa/carbidopa (4:1) was started at age 11 to 13 months, with target dosing of 5 to 6 mg/kg levodopa per day; during long-term follow-up, self-injury was apparently prevented in two patients (over follow-up of 13-14+ years) with HPRT1 gene mutations that had been invariably associated with self-injury previously (174). Future self-injury was deemed unlikely because only 1.1% of 264 published cases had self-injury onset later in life than these patients ages at the time of the report. The third patient started self-injury at age 1.5 years, while on a substantially lower levodopa dose.
Anecdotally, several other medications were reported to reduce self-injurious behavior, including atypical antipsychotics (eg, risperidone) (05), carbamazepine (141), and gabapentin (113).
Oral supplementation with S-adenosylmethionine has been introduced as a treatment for self-injury and aggression (60; 38; 21; 144). Theoretically, S-adenosylmethionine (SAMe) has therapeutic potential to replenish the brain purine nucleotide pool. Following a favorable case report on the purported benefits of SAMe in lessening self-injury (60), two small, institutional-review-board approved, open-label trials were conducted (38; 21). Both were "add-on" dose-escalation trials in which SAMe was used in conjunction with current medications. In one study, oral supplementation in five patients for at least a year (at maximum doses from 21 to 39 mg/kg per day) produced a substantial reduction in self-injury and aggression and a milder reduction of dystonia (21). In the other study of 14 patients (11 classic cases and three variant cases aged 18 to 49 years), only four tolerated the drug and reported beneficial effects, whereas seven discontinued in the first 2 months because of increased anxiety and excitability, and three experienced worsening of self-injury and increased excitement that prevented dose escalation (38).
A double-blind, 3-period, crossover trial of a single dose of the D1 dopamine-receptor antagonist ecopipam in 10 patients with Lesch-Nyhan disease was terminated early because of side effects when the drug was administered without a titration phase (93).
Enzyme replacement therapy. Two patients received enzyme replacement therapy by partial exchange transfusions every 2 months for 3 to 4 years (41); Erythrocyte HPRT activity was maintained at 10% to 70% of normal during this period, but no reduction of neurologic or behavioral symptoms was apparent.
Bone marrow transplantation. Bone marrow transplant has not been demonstrated to be either safe or effective for Lesch-Nyhan disease; it remains an experimental and potentially dangerous procedure in this condition.
A bone-marrow transplant was carried out in the United States in a 22-year-old patient with Lesch-Nyhan disease, supposedly justified on the possibility that the CNS damage is produced by a circulating neurotoxic substance of low molecular weight (122). Although the transplantation restored HPRT enzyme activity in the patient's peripheral blood cells, no change in neurologic symptoms or behavior resulted. PET scanning before and after the bone marrow transplant using a D2-dopamine-receptor ligand demonstrated no changes in receptor density following the transplantation.
Because it might be too late to reverse the cerebral symptoms in an adult, a second bone-marrow transplantation was attempted in Germany in a 16-month-old boy who had begun to show signs of psychomotor delay (43). Neither this patient nor two other affected preschool children on whom bone marrow transplantation was performed survived the procedure.
Wojcik employed a mouse model of Lesch-Nyhan disease to assess the efficacy of bone marrow transplant in ameliorating the decrease in striatal dopamine levels and behavioral hypersensitivity to amphetamine. Marrow-ablated adult HPRT-deficient mice were transplanted with marrow from congenic HPRT-expressing mice. Bone marrow transplant altered neither the neurochemical nor the behavioral phenotypes in either HPRT-expressing or HPRT-deficient mice (183).
Gene therapy. Due to the lack of success with enzyme replacement therapy and bone-marrow transplantation, the HPRT1 gene should be targeted to cells in the CNS to address the neurologic and behavioral symptoms in Lesch-Nyhan disease (155). This approach was explored by infecting mice in vivo by direct intracranial injection of a recombinant Herpes simplex type 1 virus vector containing the human HPRT1 gene (134); expression of human HPRT1 mRNA transcripts was detected in the brains of these animals. Gene therapy is not presently available, and prospects for gene therapy remain limited (106; 189). However, cell culture studies have suggested the possibility that CRISPR-mediated base editors and prime editors are a potential therapeutic strategy (84).
Surgical management. Issues addressed in a case series of nine patients with Lesch-Nyhan disease treated for orthopedic problems included (1) hip subluxation or dislocation (9 of 18 hips), fractures (n=3), infections (n=3), contractures, autoamputation, and minor scoliosis (162). Surgery results were satisfactory and resembled those of patients with spastic cerebral palsy. All seven patients who had hip surgery maintained good reduction after an average of 6-years of follow-up. With adequate cast technique, fractures, hip subluxation, and dislocation were treated successfully. Complicating factors for orthopedic management of patients with Lesch-Nyhan disease include infection, heterotopic ossification, hardware failure, and femur fracture if appropriate immobilization is not carried out.
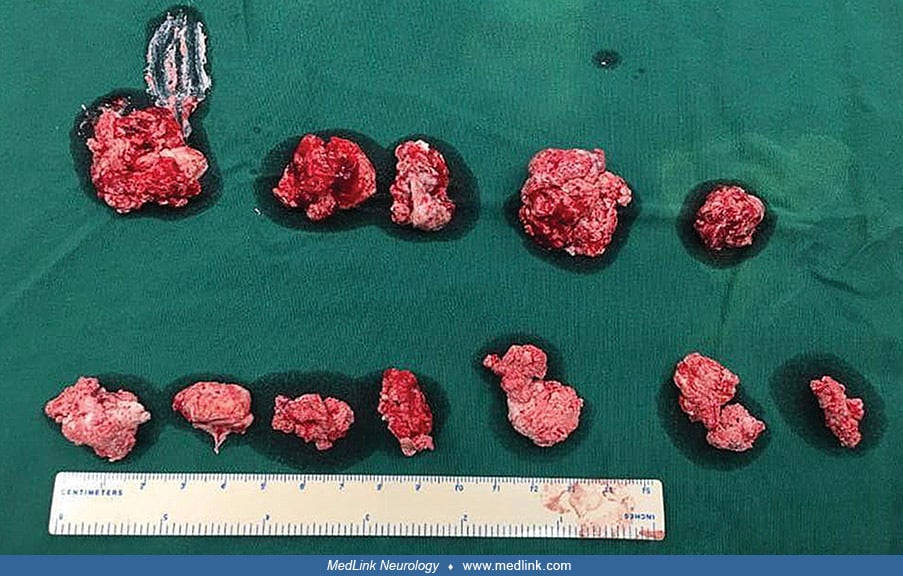
Tophi in Lesch-Nyhan disease may require bone biopsies for definitive diagnosis; bone biopsies may show urate crystals and demineralization. Tophi that rupture, interfere with joint use, or are evidently painful may require surgical debridement (06).
Bone biopsy revealed the presence of urate crystals (arrow). Hematoxylin and eosin staining. 100x magnification. (Source: Ambarsari CG, Cahyadi D, Sari L, et al. Late diagnosis of Lesch-Nyhan disease complicated with end-stage ...
Bone biopsy revealed the presence of urate crystals (arrow). Hematoxylin and eosin staining. 100x magnification. (Source: Ambarsari CG, Cahyadi D, Sari L, et al. Late diagnosis of Lesch-Nyhan disease complicated with end-stage ...
Hematoxylin and eosin staining. 100x magnification. (Source: Ambarsari CG, Cahyadi D, Sari L, et al. Late diagnosis of Lesch-Nyhan disease complicated with end-stage renal disease and tophi burst: a case report. Ren Fail 2020;4...
(Source: Ambarsari CG, Cahyadi D, Sari L, et al. Late diagnosis of Lesch-Nyhan disease complicated with end-stage renal disease and tophi burst: a case report. Ren Fail 2020;42(1):113-21. Creative Commons Attribution License. h...
Palmar view. (Source: Ambarsari CG, Cahyadi D, Sari L, et al. Late diagnosis of Lesch-Nyhan disease complicated with end-stage renal disease and tophi burst: a case report. Ren Fail 2020;42(1):113-21. Creative Commons Attributi...
Dorsum view. (Source: Ambarsari CG, Cahyadi D, Sari L, et al. Late diagnosis of Lesch-Nyhan disease complicated with end-stage renal disease and tophi burst: a case report. Ren Fail 2020;42(1):113-21. Creative Commons Attributi...
(Source: Ambarsari CG, Cahyadi D, Sari L, et al. Late diagnosis of Lesch-Nyhan disease complicated with end-stage renal disease and tophi burst: a case report. Ren Fail 2020;42(1):113-21. Creative Commons Attribution License. h...
Lithotripsy or surgery may be required for renal stones that form despite treatment (85).
Deep brain stimulation. There are now multiple anecdotal reports of improvement or resolution of self-mutilating and aggressive behavior following bilateral chronic stimulation of the globus pallidus internus for control of dystonic movements (164; 136; 23; 35; 01; 165; 175; 34; 191). Children as young as 7 years of age have been successfully treated for dystonia and self-injury with deep brain stimulation (35). Results have generally been more robust effects on self-injurious behavior than dystonia (191). When improvement in injurious or aggressive behaviors occurred, it was typically gradual over several months. In one case the parents refused a recommendation to temporarily turn off the stimulation to see if the effects were reversed (164). In another case, the behaviors returned when the stimulation was turned off but subsequently again resolved with the resumption of stimulation (23). In a 15-year-old boy who underwent bilateral placement of globus pallidus internus and deep-brain stimulation electrodes for the treatment of generalized dystonia, self-mutilating behavior gradually disappeared several weeks after the start of stimulation but returned when the right lead was fractured (01). Although deep brain stimulation appears to be a promising treatment for both the motor and behavioral disorders of patients with Lesch-Nyhan disease, the reported results have varied widely, especially for motor outcomes (34).
Sudden death. Sudden death may occur despite comprehensive case management. Although there are several causes, respiratory events are common. Factors that may cause or contribute to sudden death in these patients include aspiration, laryngospasm, central apnea, and high cervical spine injury (118). Episodes of respiratory compromise and chest imaging results may identify a need for additional tests, including potentially tracheal endoscopy, polysomnography, EEG, and imaging of the chest, brain, or cervical spine.
Patients with Lesch-Nyhan disease often require general anesthesia for dental procedures, diagnostic tests requiring immobility (eg, imaging studies), and more invasive procedures and surgeries.
Various factors complicate the use of anesthesia in this population, including increased risks of vomiting and aspiration. In addition, patients with Lesch-Nyhan disease are susceptible to severe bradycardia, apnea and other respiratory abnormalities, and sudden, unexplained death (180).
Although various anesthetic agents have been used in these patients, propofol has been advocated because of its rapid emergence and antiemetic effects (180). However, propofol anesthesia using target-controlled infusion does not provide significant clinical advantages in rapid emergence from anesthesia and management of hyperuricemia (99).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Douglas J Lanska MD MS MSPH
Dr. Lanska of the University of Wisconsin School of Medicine and Public Health has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neurogenetic Disorders
Jun. 01, 2026
Neurogenetic Disorders
May. 08, 2026
Neurogenetic Disorders
Apr. 30, 2026
Neuropharmacology & Neurotherapeutics
Apr. 23, 2026
Neurogenetic Disorders
Apr. 14, 2026
Neurogenetic Disorders
Apr. 14, 2026
Neurogenetic Disorders
Apr. 14, 2026
Neurogenetic Disorders
Apr. 14, 2026