





Neuromuscular Disorders
Viral and retroviral myositis
Jun. 16, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Congenital myotonic dystrophy is a multisystem disorder characterized by hypotonia, muscle weakness, respiratory insufficiency, feeding issues, and joint contractures in the neonatal period. Most cases are maternally transmitted due to an abnormal trinucleotide (CTG) repeat expansion of the DMPK gene. In this article, the author reviews the clinical features of children with congenital myotonic dystrophy. Extramuscular manifestations, including cardiac arrhythmias, cognitive impairment, gastrointestinal disorders, and maladaptive behaviors, are important challenges for these children. Affected individuals who survive beyond the infancy period generally experience an improvement in their motor function during childhood. Natural history studies and early results from clinical trials offer hope of new disease-modifying therapies for congenital myotonic dystrophy.
|
• Myotonic dystrophy type 1 is an autosomal dominant disorder due to abnormal expansion of trinucleotide repeats in the DMPK gene on chromosome 19q13.3. | |
|
• The severe neonatal form of myotonic dystrophy type 1 is usually associated with 1000 or more CTG repeats, with the mother being the affected parent in the majority of cases. | |
|
• Infants with the severe neonatal form of myotonic dystrophy type 1 present with hypotonia, generalized weakness, respiratory insufficiency, and feeding difficulties at birth. | |
|
• Other potential long-term complications include developmental delay, learning disabilities, behavioral problems, and intellectual disability. | |
|
• A multidisciplinary team approach is needed to address the supportive needs of affected children and their families. |
In 1901 a rare, severe neonatal form of the already recognized disease of myotonic muscular dystrophy was first described by Gardiner (30). The clinical presentation was better defined in the 1960s and 1970s by several authors and was distinguished from the more common, slowly progressive disease of the older child and adult, though occurring in the same families (91; 20; 71; 95; 07; 22; 09; 33; 83; 78). Harper pointed out that in 94% of affected neonates of both sexes, the mother was the transmitting parent despite autosomal dominant inheritance that theoretically should not be gender-specific. Additional genetic studies in the early 1990s clarified the reason as a large number of trinucleotide repeats in the defective gene of myotonic dystrophy type 1.
Affected infants have clinical problems such as reduced fetal movements and ventriculomegaly before birth, and are overtly symptomatic after delivery. Polyhydramnios is a common complication because of inadequate fetal swallowing of amniotic fluid. The most frequent clinical manifestations at birth are congenital contractures ranging from simple equinovarus deformities of the feet to arthrogryposis multiplex congenita involving the lower more than the upper limbs, hypotonia, facial diplegia with characteristic inverted V-shaped upper lip, temporalis wasting, and generalized muscle weakness.
The characteristic facies and shape of the mouth may be the first clues to the diagnosis, but this is not pathognomonic and is shared with some congenital myopathies. The palate is often high and may even be cleft.
Dysphagia is common, and infants usually require gavage and eventually may require gastrostomy. Even infants who do not have frank dysphagia may feed slowly and poorly because of the facial weakness that results in weak sucking and drooling.
Respiratory insufficiency occurs in a significant proportion of neonates with congenital myotonic dystrophy, and these infants may require supplementary oxygen, positive airway pressure ventilation, or, in some cases, tracheostomy and chronic ventilatory support (09; 78). A hemidiaphragm may be demonstrated to be nonfunctional (78).
Electrocardiographic abnormalities are found in a minority and are usually conduction defects rather than cardiomyopathy. This cardiac abnormality is usually silent, but arrhythmias and transient complete atrioventricular block during the neonatal period are possible (43).
The testes of male infants often are undescended.
Congenital cataracts are infrequent and often require a slit-lamp examination to be detected early.
A serious complication of neonatal myotonic dystrophy is poor peristalsis due to smooth muscle involvement (78). The abdomen becomes distended from air in the stomach and intestines and may push up against the diaphragm, further compromising respiration. Obstipation and inability to pass stool, or even the meconium plug initially, is a difficult complication.
Poor muscle function of the gallbladder may also produce cholelithiasis or cholestasis (81).
Endocrine abnormalities may involve the thyroid and adrenal medulla for cortisol production as well as regulation of blood sugar because of insulin resistance; vigilance for hypoglycemia is needed.
Seizures are not a feature of neonatal myotonic dystrophy unless the infant has hypoxic-ischemic encephalopathy. Intrapartum asphyxia is a serious problem in infants delivered prematurely, and the risk of germinal matrix and intraventricular hemorrhage is greater than in preterm infants of comparable gestational age that do not have myotonic dystrophy (59). The MRI in congenital myotonic dystrophy often shows ventriculomegaly and hyperintensity of white matter posterior and superior to the trigone region, with no correlation to age or trinucleotide repeat size (46; 62).
Prognosis is guarded for infants who have respiratory insufficiency and dysphagia, and many die early. Affected infants requiring ventilation support for more than 30 days had a 25% risk of mortality during the first year (14); furthermore, those with preterm delivery requiring resuscitation or therapeutic hypothermia at birth, or those who had severe hypoxic-ischemic encephalopathy were associated with reduced survival (63). Survivors generally experience an improvement in muscle strength and motor function during first decade of life (72), reaching a plateau during adolescence, and then decline after the second decade (45). Dysarthria is a frequent complication among children with congenital myotonic dystrophy due to impaired orofacial functioning (80; 08). These children may also have other chronic health issues related to gastrointestinal dysmotility, sphincter dysfunction, myotonia, cardiac arrhythmias, respiratory insufficiency, skeletal deformities, and cataracts (47). Difficulties with communication, fatigue, process and motor skills are frequent symptoms that negatively impact their activities of daily living and health-related quality of life (40; 25). Learning disabilities and cognitive impairment are common among children with congenital myotonic dystrophy (17; 82; 67). These individuals may display maladaptive behaviors that further restrict their social participation long term (28). In a longitudinal study, the gap in cognitive and adaptive functioning among children with congenital myotonic dystrophy type 1 widened over time compared to age-matched healthy controls (49). Furthermore, children and adolescents with myotonic dystrophy type 1 are at increased risk for neuropsychiatric comorbidities such as autism, anxiety, and attention deficit disorders (23; 24; 21; 47; 66). In a study using United States insurance claims data from 2012 to 2019, the mean costs of healthcare resource utilization and all-cause healthcare expenses were found to be highest ($66,496 vs. $2828 USD) for patients with congenital myotonic dystrophy during the first 12-month follow up; their mean costs remained markedly elevated (above $17,944) across all subsequent period (up to 47-month) when compared to age matched controls due to their significant comorbidities (37).
A 1-day-old infant girl was referred for neurologic assessment because of multiple joint contractures, respiratory distress, and feeding difficulties. The pregnancy was by in vitro fertilization due to previous maternal miscarriages, and reduced fetal movement was noted antenatally. Cesarean section was performed at 36 weeks because of breech presentation. Her birth weight was 1.81 kg, and Apgar scores were 4 at 1 minute and 7 at 5 minutes. Mild dysmorphic features were noted, including down-sloping palpebral fissures, low-set ears, elevation of the palate, and short neck. She had bilateral talipes equinovarus feet, dislocated hips, and adducted thumbs. Aside from a grade 3/6 pansystolic ejection murmur, the remaining general examination was normal. Her neurologic examination revealed truncal hypotonia, reduced muscle bulk, diminished deep tendon reflexes, and diffuse muscle weakness. Her cranial nerves showed reduced facial expression and diminished gag response, with no tongue fasciculations or cataracts. She required supplemental oxygen during the first week and was gavage-fed until 3 weeks of age.
Examination of the mother was positive for distal hand weakness and grip myotonia, which had been noted since adolescence. She was otherwise in good health. The maternal grandfather had surgery for cataracts in his 50s and developed type II diabetes.
Subsequent investigations in this infant revealed mildly elevated creatine kinase of 461 U/L. Her serum glucose and thyroid functions were normal, as was her initial chest and abdominal x-ray. EKG showed normal sinus rhythm with left axis deviation and right ventricular hypertrophy. A small atrial and ventricular septal defect was noted on echocardiogram. Karyotype and newborn metabolic screen were normal. Molecular genetic testing showed 700 CTG trinucleotide repeats of the DMPK gene in the infant and 200 repeats in the mother.
Myotonic dystrophy type 1 is a multisystem autosomal dominant disorder due to abnormal expansion of CTG trinucleotide repeats in the 3’-untranslated region of the dystrophia myotonica protein kinase (DMPK) gene on chromosome 19q13.3 (10; 27; 52; 84). The amount of abnormal expansion generally correlates with the severity of the weakness and other clinical manifestations (45; 98). The number of CTG repeats in normal alleles is five to 37. In the more common adult and late-onset forms of myotonic dystrophy, more than 50 to 350 repeats are detected. Earlier age at onset of disease was associated with longer repeat length and more clinical severity, although substantial variability was noted among affected individuals (16; 89). Both repeat length and prematurity were significantly associated with disease severity (76). In the severe, neonatal form of myotonic dystrophy type 1, 1000 or more repeats usually are demonstrated (73).
The CTG repeat allele on 19q13.3 is relatively stable in somatic tissues but highly unstable in the germline and extremely biased toward further expansion, which is consistent with the high level of anticipation observed in families (58). The mother is the affected parent in most cases because the gene is less stable in the maternal DNA of the ovum, and excessive repeats are, thus, more readily produced from the maternal DNA (73). Anticipation occurs more frequently in maternal (85%) than paternal (37%) transmissions (04). Paternal transmission has also been reported in rare cases of congenital myotonic dystrophy (85; 99; 18; 47). Barbé and colleagues proposed that CpG hypermethylation of an insulator protein CTCF binding (CTCF1) site flanking the CTG repeats in the DMPK gene may account for the maternal bias for congenital myotonic dystrophy transmission (06). Furthermore, the degree of CTCF1 hypermethylation is mutant allele length-dependent and may serve as a biomarker for congenital myotonic dystrophy (48; 57).
The genetic and molecular bases of the myotonic dystrophies are summarized in several reviews (55; 56; 02). In myotonic dystrophy type 1, abnormal intranuclear accumulations of ribonucleic acid (RNA) inclusions alter the activities of various RNA processing factors, including muscleblind-like (MBLN) and CUG-binding proteins (CUGBP), leading to misregulation of developmentally programmed alternative splicing (69; 50). Failure to express mature splicing transitions in targeted mRNAs and nuclear sequestration of muscleblind-like proteins impairs myogenic differentiation and leads to other cellular deregulation in various tissues; the degree of alternative splicing and polyadenylation abnormalities is significantly more extensive and occurs prenatally in congenital myotonic dystrophy type 1 (86; 32). Accumulation of mutant pre-mRNA and aberrant alternative splicing of a number of genes in the cortical neurons, including the microtubule-associated tau protein, the amyloid precursor protein, the SLITRK family of proteins, and the N-methyl-D-aspartate receptor 1, contributes to central nervous system disease in myotonic dystrophy type 1 (12; 31; 61). Other factors, including abnormal repeats-associated translation, activation of protein kinase C-dependent signaling pathway, aberrant polyadenylation, and microRNA deregulation may also contribute to the complex pathogenic mechanisms (15; 17; 32).
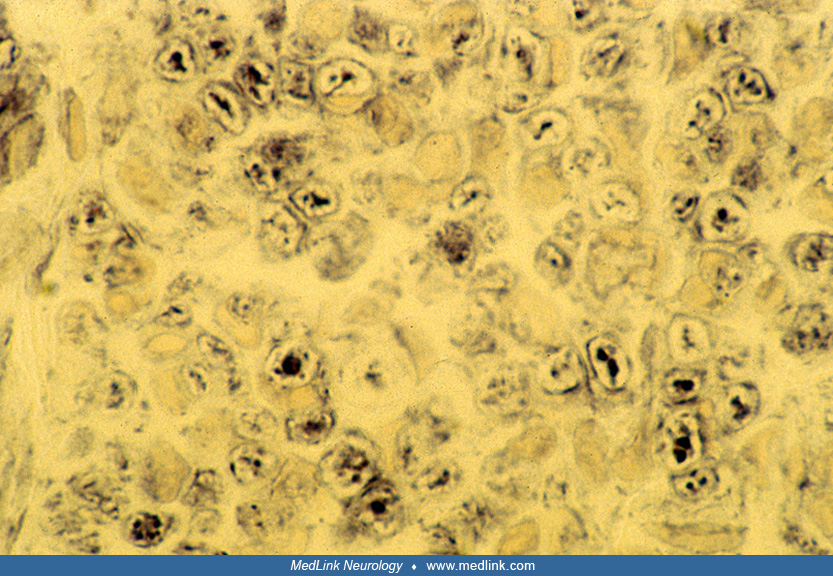
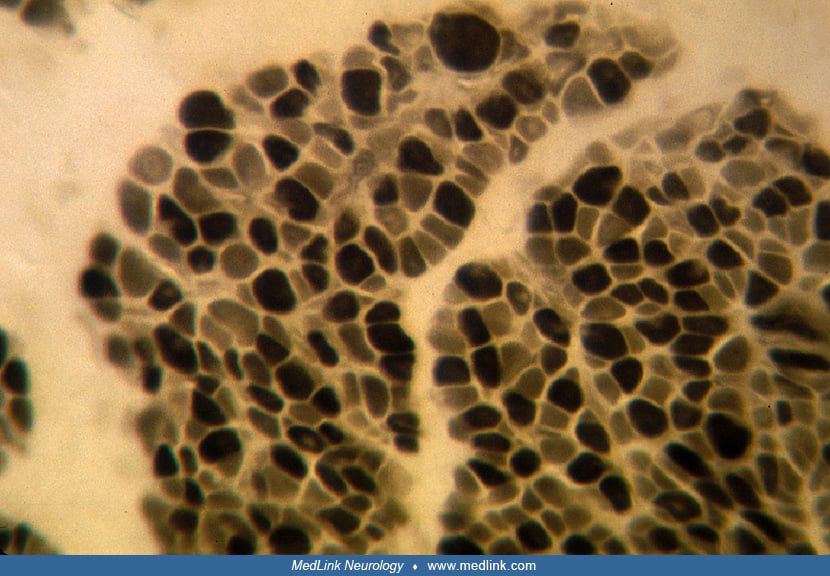
The cause of striated muscle weakness in the neonatal period and early infancy is muscular immaturity rather than necrosis or degeneration. The muscle biopsy in neonatal myotonic dystrophy shows variable degrees of immaturity, with myoblasts, myotubes, undifferentiated myofibers, and mature myofibers all mixed randomly within the same fascicles; ultrastructural evidence of maturational delay corroborates the histological and histochemical findings (42; 26; 79).
Persistent fetal expression of vimentin and desmin intermediate filaments also is demonstrated (77). Correlation between the number of trinucleotide repeats and the severity of maturational delay in muscle is poor, however (84). Another pattern of the muscle biopsy sometimes found in congenital myotonic dystrophy is congenital muscle fiber-type disproportion, either fully or partially developed, as uniform smallness and more than 80% predominance of type I myofibers (03; 88).
The muscle in neonatal myotonic dystrophy, unlike that of X-linked myotubular myopathy, is a true maturational arrest; the appearance of the biopsies in these two diseases is different and can be distinguished histologically. The absence of degenerating and regenerating myofibers and of connective tissue proliferation distinguishes neonatal myotonic dystrophy from congenital muscular dystrophy and the other form of muscular dystrophies.
Smooth muscle also is extensively involved in myotonic dystrophy and causes problems with gastrointestinal motility in the neonate and abnormal uterine contractures during labor in the pregnant mother (78).
The reported incidence of congenital myotonic dystrophy ranges from 1 in 3500 to 100,000 live births (96; 13). All ethnic and racial groups are affected.
Genetic counselling should be offered to individuals with a family history of myotonic dystrophy, and especially to those with confirmed DMPK mutation (41; 89). Prenatal diagnosis is available from chorionic villus samples and cultured amniocytes, from which DNA analysis can be performed during the first half of gestation at 8 to 20 weeks. Fetal cord blood may be obtained in older fetuses and genetic studies performed on leukocytes. The analysis of DNA from these samples shows accuracy in CTG repeat size, and when combined with DNA methylation studies can help predict severe neonatal disease (73; 94). Trinucleotide repeats contraction is a pitfall to be aware of in the interpretation of genetic testing for prenatal diagnosis (01; 54). The frequency of contractions may be higher in DM1 families with variant (non-CTG) expansion repeats; long-read sequencing with assessments of the expansion size, the degree of somatic instability, as well as the pattern of variant repeats and DNA methylation may provide a more comprehensive understanding of disease severity in patients with myotonic dystrophy type 1 (68).
The neonatal form of myotonic dystrophy should be differentiated from congenital muscular dystrophy, congenital myasthenia gravis, spinal muscular atrophy, mitochondrial cytopathy, severe congenital myopathies, amyoplasia, and other causes of arthrogryposis multiplex congenita.
Neurologic examination of the parents, particularly of the mother, is essential. The single most important confirmatory diagnostic test is the molecular genetic marker in the blood for myotonic dystrophy type 1, which will also provide the number of trinucleotide repeats (98).
EMG is not helpful because myotonia is not yet developed at birth, and the findings are nonspecific. Muscle biopsy is now rarely indicated.
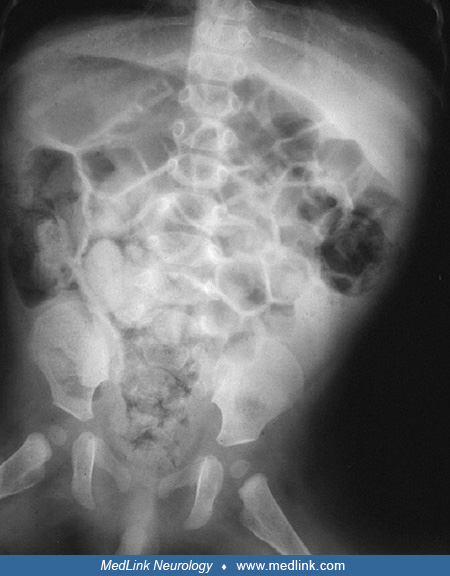
Roentgenograms of the chest and abdomen are used to help determine the status of diaphragmatic and gastrointestinal functions. Thin ribs are frequently demonstrated, as in other severe neuromuscular diseases beginning in fetal life. Ultrasound or fluoroscopy may be needed to further define diaphragmatic hemiparesis. A swallow study may be indicated for infants with feeding difficulties; close monitoring is required to detect early respiratory insufficiency. An electrocardiogram (EKG) should also be performed for all infants with myotonic dystrophy, even if asymptomatic; echocardiography and Holter monitoring may be required in subsequent follow up evaluations (93; 11). Brain MRI often reveals white matter disease, enlarged ventricles, cortical atrophy, and other nonspecific brain abnormalities (19; 31; 62; 17).
The eyes should be examined with a slit-lamp for congenital cataracts; standard direct ophthalmoscopy is not adequate. Endocrine evaluations, including serum cortisol, thyroid function, HgA1C, blood glucose, and lipid profile, should be performed periodically. Serum creatine kinase is nondiagnostic and may be normal.
In the neonatal period, the most immediate needs are for respiratory and feeding support. The diagnosis must be confirmed. Attention to cardiac status and GI motility are important. Ventriculomegaly is common, but most cases will not require shunting (60). Arthrogryposis may require surgical correction of the most severe contractures, and physiotherapy should be started early. Early speech therapy referral is indicated to help with communication challenges (40; 80). Periodic neuropsychologic assessments can help determine the degree of cognitive impairment (65). Gastrointestinal and urological symptoms are common and often require treatment (39; 51). A multidisciplinary approach including psychosocial therapy is necessary to optimize the health outcomes of these children and prepare them for transition to adult services (29; 36; 05; 35; 39).
Emerging treatments including gene therapy are being evaluated for adults with myotonic dystrophy type 1 (44; 31; 70; 87; 53; 34). AMO-02, an inhibitor of an intracellular regulatory kinase GSK3B, was reported to have a favorable clinical profile in a clinical trial involving adolescents and adults with congenital and childhood-onset myotonic dystrophy type 1; further trial is on-going (38). Future therapies will hopefully ameliorate both the muscular and extramuscular manifestations of this disease (64; 74).
A high rate of fetal loss occurs due to spontaneous abortion, and premature delivery is frequent. In fetuses carried to term, fetal movements are reported by mothers to be less than in her other pregnancies with nonaffected infants. Maternal complications of abnormal labor due to poor uterine contractions, placenta previa, and retained placenta are several times more frequent in mothers with myotonic dystrophy than in the normal population (78; 97). Mothers with a known or suspected diagnosis of myotonic dystrophy type 1 should receive prenatal care by a high-risk obstetrician familiar with these potential complications. As well, a pediatrician or neonatologist should be present at delivery to provide necessary supportive care (39).
There are no specific contraindications, but as with any congenital myopathy, the risk is high for apnea and respiratory failure after the anesthetic. Careful perioperative planning is required to minimize complications (92; 75; 90).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.
Jean K Mah MD
Dr. Mah of the University of Calgary has no relevant financial relationships to disclose.
See Profile
Harvey B Sarnat MD FRCPC MS
Dr. Sarnat of the University of Calgary has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neuromuscular Disorders
Jun. 16, 2026
Neuromuscular Disorders
May. 27, 2026
Neuromuscular Disorders
May. 21, 2026
Developmental Malformations
May. 08, 2026
Developmental Malformations
May. 08, 2026
Developmental Malformations
May. 08, 2026
Developmental Malformations
May. 08, 2026
Neuroimmunology
Apr. 26, 2026