






Developmental Malformations
Barth syndrome
May. 08, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
|
• Anterior pituitary aplasia or dysplasia is associated with endocrinopathies and often with malformations of the eyes, brain, or somatic anomalies; multiple genes are implicated, particularly LHX4, OTX2, and HESX1. | |
|
• Ectopic neurohypophysis and posterior pituitary dysplasia are mainly associated with anterior pituitary endocrine defects because of early neural induction of the Rathke pouch; the HESX1 gene is mostly implicated, but multiple genes are implicated in both isolated and syndromic cases. | |
|
• Pituitary aplasia and dysplasias may occur as isolated anomalies or as part of major brain malformations, most frequently with septo-optic dysplasia, holoprosencephaly, and Joubert syndrome. Duplications of the pituitary can also occur. Neuropathological findings are variable. | |
|
• Clinical manifestations are largely related to the endocrinopathies but also may involve intellectual deficits, visual problems, and olfactory impairment. | |
|
• Diagnosis is based on clinical history, physical examination, endocrine function studies, and neuroimaging, particularly MRI. | |
|
• Treatment addresses the correction of endocrine deficiencies. | |
|
• Growth hormone deficiency is the most frequent endocrinopathy of infancy, but other pituitary hormonal insufficiencies may appear later in childhood, so vigilant monitoring is recommended in follow-up. | |
|
• Numerous genes are reported in isolated growth hormone deficiency, but most frequently due to GH1 and GHRHR mutations. |
Pituitary aplasia (absence) is a rare condition first described in the 1950s and 1960s (37; 192; 228). It has been linked to ciliary gene mutations, as exemplified in patients with type 9 oral-facial-digital syndrome (03).
Pituitary hypoplasia, in which the pituitary gland is diminutive and leads to congenital hypopituitarism, is not always an anatomical anomaly of the adenohypophysis or neurohypophysis (anterior and posterior pituitary) identified in neuroimaging studies to correlate with functional neuroendocrine disorders. Ectopic (ie, displaced) neurohypophysis is a common anatomical anomaly, with or without other brain malformations.
The term pituitary dysplasia may be associated with abnormalities in the adenopituitary cells (109; 199; 140), abnormalities in the neuropituitary cells (145; 18), or other abnormalities in the tissue components that are vital for the organ. Ectopic adenopituitary tissue was found pharyngeally (163), and ectopic neuropituitary tissue has been described in cases where the neuropituitary gland is either absent or underdeveloped (14; 47).
The diagnoses of pituitary aplasia, hypoplasia, and dysplasia are difficult to make. In the diagnostic process, three principal methods are used: (1) endocrinological, (2) radiological, and (3) autopsy methods. In extremely preterm neonates, serum hormone levels differ significantly from those of late-preterm and full-term neonates; therefore, it is important that the hospital laboratory has reference concentrations at different gestational ages of preterm infants (90).
The diagnosis of pituitary agenesis (the absence of pituitary gland primordium) can be made only after the pharyngeal side of the cranial base has been examined for adenohypophyseal (anterior pituitary) tissue, and the brain has been investigated for neurohypophyseal (posterior pituitary) tissue by MRI or neuropathological examination.
The diagnosis of pituitary aplasia, in which no pituitary gland tissue is found where the gland is normally located in the sella turcica, necessitates a description of the morphology of the sella turcica. In abnormal sella turcica morphology (such as that observed when a craniopharyngeal canal has formed in the floor of the sella turcica), glandular tissue may be found in the osseous canal (126; 130; 129). Similar precautions are necessary in order to make the diagnosis of hypoplasia and dysplasia. The nomenclature of pituitary abnormalities should take into account the complicated normal embryological origin of the gland (41). The sella turcica, a superior depression in the sphenoid bone, also has a complex embryology and is altered in size and shape with developmental structural defects of the pituitary (235).
In this review, the term “pituitary aplasia” will be used to refer to the condition observed when the pituitary gland is absent or when a rudimentary gland in a normally developed sella turcica is recorded. Accordingly, this condition does not include cases in which pituitary tissue is found outside the sella turcica.
A frequent association of pituitary dysfunction is septo-optic-pituitary dysplasia. This disorder should be considered a clinical syndrome rather than a neuropathological entity because of the wide range of variability in neuropathological findings and genetic mutations (202; 207). Multiple comorbidities may occur in patients with optic nerve hypoplasia and septo-optic-pituitary dysplasia (15).
“Pituitary dysplasia” is understood to be all forms of defect in the organ caused by defective development, including malposition of pituitary tissue and ectopic neurohypophysis. The “pituitary stalk interruption syndrome” is characterized by a triad of interrupted pituitary stalk, absent or ectopic neurohypophysis, and anterior pituitary hypoplasia or aplasia, the latter often leading to panhypopituitarism; the diagnosis is confirmed by MRI (190; 248). This triad is really just a variant of the ectopic neurohypophysis syndrome.
Rarely, by contrast with agenesis, the pituitary may be duplicated (44; 86). Hyperplasia of the single midline pituitary may occur in hypothyroidism (254).
Congenital pituitary aplasia and dysplasia is a rare disorder that most often occurs in combination with other midline craniofacial abnormalities, such as septo-optic aplasia, anencephaly, and holoprosencephaly (13). When associated with septo-optic dysplasia, other variable anomalies may be associated, including congenital oculomotor palsy (265) and anomalous cerebral vessels, including anomalous origin of the anterior choroidal arteries, absence of the great vein of Galen, and right Rosenthal vein leading to the superior petrosal sinus (46). Neonates with septo-optic-pituitary dysplasia may show hypernatremia, hypoglycemia, and persistent hypothermia (27). Septo-optic-pituitary dysplasia is the most frequent cause of optic nerve hypoplasia, and most patients have hormonal deficiencies (246). Rarely, hypopituitarism also may include prolactinoma (189). The degree of optic nerve hypoplasia on MRI in septo-optic-pituitary dysplasia and the severity of clinical visual impairment are not always concordant (204). Optic nerve hypoplasia is usually bilateral but may be asymmetrical or unilateral (171). Aplasia of the olfactory bulbs, tracts, and sulci occurs in 8.6% of cases (204). Callosal agenesis or hypoplasia occurs in 15% of patients with septo-optic-pituitary dysplasia (171).
Panhypopituitarism is the general finding in pituitary aplasia and dysplasia (30; 169). Signs and symptoms of anterior hypopituitarism are almost always present, whereas posterior pituitary function may or may not be impaired. Accordingly, growth hormone is almost always deficient, whereas adrenocorticotropin and thyroid-stimulating hormone are deficient in a large proportion of patients. Clinical deficiencies of the other adenohypophyseal hormones are less common. There may be progression from isolated growth hormone deficiency to multiple pituitary hormonal deficiencies in some patients (04). The CHARGE syndrome may be associated with structural abnormalities of the pituitary and panhypopituitarism (91). Other rare associations of pituitary hypoplasia include Rubinstein-Taybi syndrome, in which some patients also exhibit a mutation in a novel cAMP-binding protein (157).
Normally, several symptoms of hypopituitarism present early in the neonatal period. These include sluggishness, lethargy, cyanosis, shock, and other nonspecific symptoms presenting at birth or developing within hours thereafter. Severe hypoglycemia, frequently associated with seizures, is a key symptom that generally appears during the first day of life. Hypoglycemia is seen in 15% to 35% of patients. Neonatal hyponatremia and persistent hypothermia may accompany the hypoglycemia (27).
Male infants show underdevelopment of the external genitalia in the form of microphallus, undescended testes, or underdeveloped scrotum, whereas the external genitalia of females are unaffected (119).
The disorder is often associated with hypoplastic thyroid and adrenal glands, along with hypothyroidism and adrenocortical insufficiency. Hypothyroidism is not revealed clinically during the first days of life; therefore, the results of neonatal thyroid screening programs may be the first recognized sign of this endocrine malfunction. If undiagnosed, the adrenocortical insufficiency may be the cause of death (236).
Hypopituitarism in pituitary aplasia and dysplasia manifests earlier than in children with acquired pituitary lesions. Growth hormone is not essential for intrauterine growth, and the infants are of normal length and body weight at birth. Noticeable growth retardation is present at 12 to 18 months of age and may be present as early as 3 to 6 months of age. The body proportions are normal (75). The patients tend to be obese. The patients’ height-age is less than their weight-age. The excess fat is frequently concentrated in the chest and abdomen. Delayed tooth eruption and a round face are usually found. Neurodevelopment (cognitive, psychosocial, and language skills) is generally delayed in untreated children; however, this is much less so when children are diagnosed early and appropriately treated.
Coupled with the endocrinological manifestations are manifestations of impaired skeletal development. Skull x-ray, CT scans, and MRI scans may reveal an empty sella or a craniopharyngeal canal (93).
An association between hypopituitarism and unilateral agenesis or hypoplasia of the internal carotid artery has been reported (142; 208). The genetic basis is unknown but is not due to mutations in the HESX1, LHX4, or OTX2 genes (142).
Hyperbilirubinemia and prolonged icterus, along with hepatitis-like histological hepatic changes, have been reported (98; 94).
Hypothalamic-pituitary dysfunction secondary to dysgenesis may result in psychiatric symptoms, particularly depression and even suicide in adolescents (34; 150; 166).
A solitary median central incisor may be a useful clinical biomarker of hypoplastic anterior pituitary and ectopic neurohypophysis and is associated with holoprosencephaly or septo-optic-pituitary dysplasia; therefore, the teeth should be observed during physical examination (65).
Isolated pituitary deficiencies are not associated with epilepsy except in septo-optic-pituitary dysplasia, in which seizures occur in only a minority of patients but more often than in age-matched control populations, and some even have epileptic (infantile) spasms initially in early infancy (216).
The cause of pituitary aplasia and dysplasia is a genetic defect that causes an early embryological developmental defect in the hypophyseal placode or in the processes that initiate the transformation of the placode into the hypophyseal organ. The suggestion has been posited that the notochord is decisive in this developmental process (130).
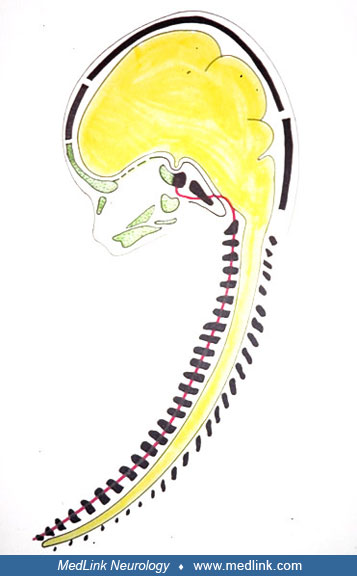
Normal ontogenesis. The embryology of pituitary development is that the adenohypophysis develops from the outpouching of ectoderm at the roof of the oral cavity (Rathke pouch) and the neurohypophysis, infundibulum, and pituitary stalk from the neuroectodermal floor of the diencephalon (102; 206; 09). The primordium of the adenohypophysis can be distinguished in the human embryo as early as 22 days gestation (68), and the HESX1 gene is implicated in this development (55). Five different cell types secreting six different hormones all arise from a common progenitor; morphogenesis involves complex regulatory networks with transcription factors and both intrinsic and extrinsic signaling molecules (09). Both neural crest cells and endoderm-lineage cells invade the developing primordial pituitary and, in the adult, contribute to maintenance and perhaps to neoplasia (116).
Rathke pouch invaginates upward from the oral cavity toward the ventral diencephalon at about 28 days, eventually making direct contact with the diencephalon in the midline, separated from it only by a thin intervening basement membrane as it elongates (227). The anatomical development of the pituitary is complete by 49 days and is followed by functional specialization and differentiation of the various hormonal secretory cells with formation of the portal circulation; reciprocal inductive signals between the diencephalon and Rathke pouch affect growth hormone (89). The growth hormone deficiency in ectopic neurohypophysis can, thus, be explained, even though growth hormone is of adenohypophysial origin. Persistence of the craniopharyngeal canal occurs with ectopic neurohypophysis secondary to SOX3 gene deletions in both mice and humans (08). Persistent expression of activated Notch in the developing hypothalamus affects the survival of pituitary progenitor cells and alters pituitary structure, but the importance of Notch in the pathogenesis of most cases of human ectopic neurohypophysis is uncertain (17). An important gene with an essential role in hypothalamic neuronal lineages in the mouse is Orthopedia (Otp), which is expressed in various nuclei, including the anterior periventricular, paraventricular, and supraoptic (249). Rare cases of infundibular aplasia associated with ectopic neurohypophysis have been described, but the genetic basis is not yet established (253). Some cases with extensive telencephalic and diencephalic malformations are attributed to a neuroblast migratory disorder (176), but the mechanisms of pathogenesis are multiple in these cases, and neuroblast migration is only one of several impaired developmental processes. Normal hypothalamo-pituitary development is closely related to that of the forebrain and depends on complex genetic cascades of transcription factors and signaling molecules either intrinsic or extrinsic to Rathke’s pouch (120). Neuroredoxin (NXN), which encodes a multi-functional enzyme with oxidoreductase activity, regulates WNT signaling during pituitary progenitor stem cell differentiation (38).
Abnormal ontogenesis. The normal embryological development of the pituitary gland is disturbed in pituitary aplasia and dysplasia (21). The embryonic region, from which the pituitary gland arises, can (analogously to the nasal, lens, and otic placodes) be termed the “pituitary placode.” All four placodes exhibit the same structural stratification in the placode region, characterized by direct adherence of surface ectoderm to neuroectoderm. This basic connection does not disappear during subsequent normal development. In the pituitary gland, this adherence occurs throughout life at the site where the intermediate part (derived from surface ectoderm) adheres to the neuropituitary gland (derived from neuroectoderm) (132). This concept of pituitary development by sustained placode adherence differs from that proposed earlier, in which an invagination of surface ectoderm gradually extended cranially and finally fused with the infundibulum. During development, the surface ectoderm and neuroectoderm both form process-like extensions (Rathke pouch and the infundibulum, respectively). These anatomic structures are presumed to arise by mesodermal "filling up" of parachordal tissue and neural crest cells around the persisting placodal contact. This means that the surface ectoderm is drawn out of the pharyngeal mucosa, from where it is disrupted, and the neuroectoderm is drawn out in an infundibular process of the diencephalon. Transcription factor 7-like 1 (TCF7L1) is involved in hypothalamo-pituitary axis development in both mice and humans, particularly for induction and subsequent expansion of Rathke pouch progenitors; its deficiency results in pituitary aplasia and sometimes forebrain anomalies (83).
After these changes in the surrounding mesoderm, the sella turcica develops, first in cartilage and then in bony tissue (128). Once the sella turcica has formed, the connection through the cranial base is severed, and the boundary between the posterior notochordally derived cranial base and the anterior (mainly neural crest-derived) cranial base can no longer be distinguished in the sella turcica. The pathogenesis and pathophysiology for maldevelopment of the pituitary gland may accordingly be molecular biological defects in (1) the surface ectoderm or in adhesions in the placode region, (2) defects in parachordal mesodermal penetration (so-called "filling up"), and (3) defects in neuroectodermal development.
In holoprosencephaly, an antenotochordally located defect is seen in the surface ectoderm, mesoderm, and neuroectoderm (219; 206). The developmental defect extends in the horizontal plane in a fanlike shape from the pituitary gland to the eyes and in the frontal plane in a fanlike shape from the premaxilla to the eyes (133; 129). This hereditary condition may often be attributable to the Sonic Hedgehog gene (196). In holoprosencephaly, a dysplastic development of the pituitary gland occurs with malposition of the adenohypophyseal tissue and malformation of the sella turcica (67; 126; 42; 160).
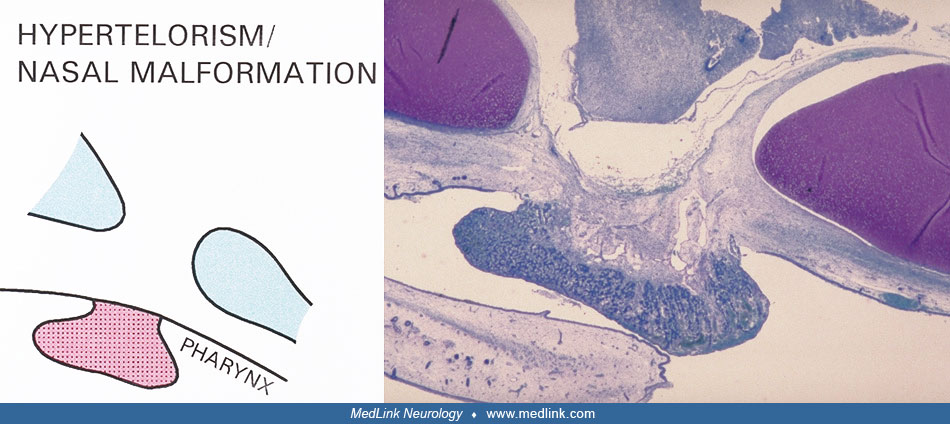
In nasal placode malformation, in which the nasal cavity does not form, hypophyseal malformation can likewise be seen, resulting in no pituitary gland primordium in the sella turcica region but with the entire adenohypophysis located in the pharynx (136). In such cases, there has likely been defective adhesion between surface ectoderm and neuroectoderm. The reason for this is not known. Earlier studies have highlighted the fact that hypopituitarism or hypothalamic dysfunction occurs in malformations other than holoprosencephaly and nasal placode malformation. These malformations include septo-optic dysplasia, midfacial anomalies (eg, hypertelorism), cleft lip and palate, Rieger syndrome, Pallister-Hall syndrome, and solitary maxillary central incisor (73; 191; 149; 198; 69; 95; 229; 105; 123; 50; 156; 168; 197; 141; 220).
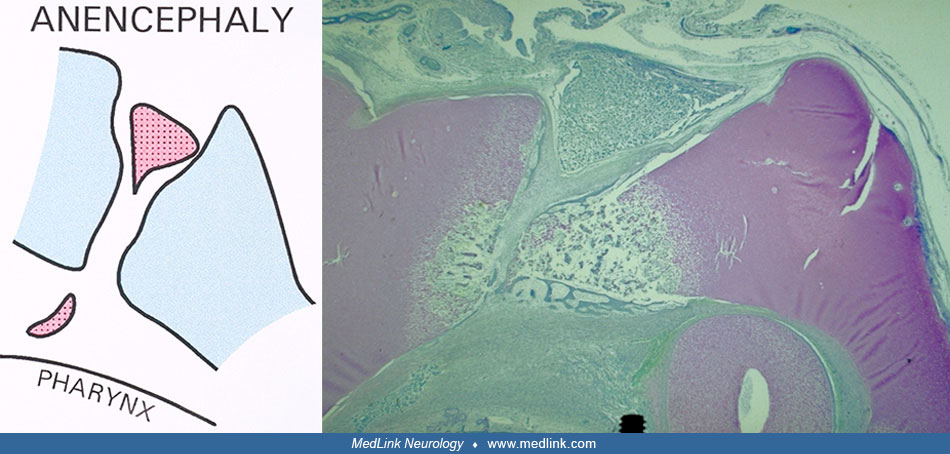
Defects in parachordal mesodermal tissue penetration and in neuroectodermal development may be the cause of pituitary dysplasia as seen in anencephaly, where the neurohypophysis is often absent and where a craniopharyngeal canal is seen in the floor of the sella turcica. The same etiology may be behind transsphenoidal encephalocele, where pituitary aplasia is observed (127). These types of malformation may be related to notochord malfunction.
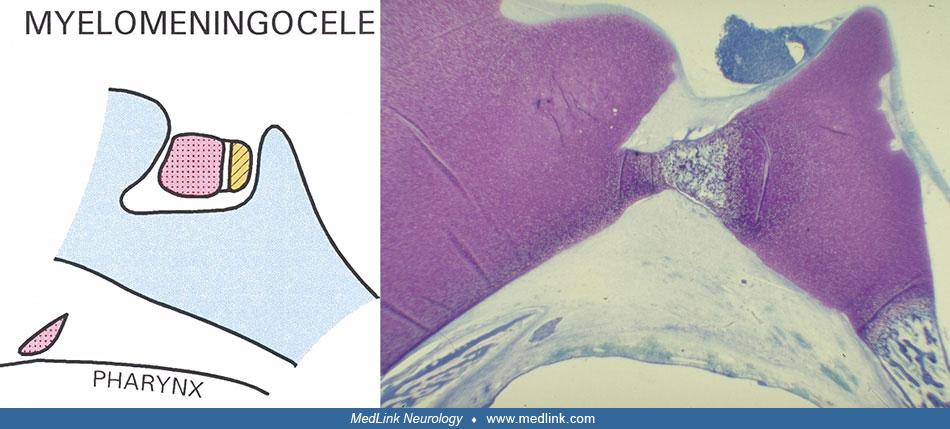
In spina bifida, ascribed to deviations in the Sonic Hedgehog gene expression (20), pituitary dysplasia is observed (131).
Consequently, the pathogenesis and pathophysiology of pituitary aplasia and dysplasia show that the sella turcica is a boundary region between the antenotochordal developmental field and the notochordal developmental field (126).
It is now possible to create in vitro pituitary organoids as experimental models (113).
Genetic bases and correlates. Animal data indicate that the pit-1 gene is necessary for the specification of the phenotype of the three cell types of the anterior pituitary gland (147). Mutations in the pit-1 gene have been demonstrated in children with combined pituitary hormone deficiency (174; 188; 77). A novel pituitary paired-like homeodomain factor, appearing to be an important prerequisite for the expression of the transcription factor pit-1, is named prophet of pit-1 (64). In a review, Westphal summarized the genes that fashion the pituitary gland in the following way (252):
|
One of the first gene products involved in early pituitary organogenesis is BMP4, a member of the TGF [beta] family of secreted signaling molecules. BMP4 is expressed in the neural ectoderm of the infundibulum. Thereafter, the infundibulum releases FGF8. FGF10 and BMP2 are additional secreted molecules that have been implicated in the process of forming the Anlage of the pituitary gland. These pituitary-specific patterns are initiated by two members of the LIM-homeobox gene family of transcription factors, termed Lhx3 and Lhx4. The next stage of pituitary development is characterized by the activity of control genes Pitx1 and Pitx2. This period of precursor expansion appears to be controlled by Rpx/Hesx1, a member of the paired-like family of homeobox genes. The formation of the hormone-secreting pituitary gland is heralded by the emergence of Prop1, another member of the paired-like family of homeobox genes. Prop1 activity is needed for the expression of Pit1. Early cell-to-cell signaling, mediated by secreted morphogens, results in the definition of organ rudiments. A missense variant in LHX4, related to GLI2 and AGFR1 variants, were reported in two unrelated families of children, each with multiple pituitary hormonal deficiencies (205). |
Sbrogna and colleagues suggest that “Hh signaling from neural ectoderm is necessary for induction and functional patterning of the vertebrate pituitary gland” (210). Ectopic hypophysis or pituitary stalk interruption syndrome is characterized by genetic heterogeneity as demonstrated by whole-exome sequencing, including rare and novel variant genes (36; 221).
Over the last few years, there has been an increasing clinical interest in correlating genetic abnormalities with the phenotypic appearance. Initially, this correlation was observed in patients with Pit-1 mutations, but subsequent analyses of other genes have been conducted, and many are now implicated (87). An example is a study of a family with an LHX4 germline splice-site mutation resulting in short stature and pituitary and hindbrain defects in combination with abnormalities of the skull base (153). Other authors do not find a strong correlation between posterior pituitary deficiency in particular and LHX2 mutations (179). Absence of the anterior pituitary with panhypopituitarism and ectopic neurohypophysis is seen in 1p31.1-1p31.3 microdeletion (237). Mutations in the BMP4 gene cause pituitary insufficiency associated with anophthalmia and polysyndactyly (222). The OTX2 gene is also implicated in some cases of pituitary dysgenesis with hormonal deficiencies associated with pineal, ocular, and ear defects (84; 222). PAX6 mutations may impair pituitary function, but aniridia or anophthalmia are more frequent expressions (218). Similar ocular anomalies with hypopituitarism and other anomalies are associated with ARNT2, SOX2, and OTX2 mutations (251; 152). Other genes involved as transcription factors in pituitary development include PROP1, POUF1, LHX3, and LHX4, but mutations of these genes have not been shown to cause pituitary aplasia (150). Optic nerve hypoplasia, a component of septo-optic-pituitary dysplasia (DuMorsier syndrome), does not necessarily predict the hypopituitarism that occurs in many but not all young patients (81), but all children presenting with optic nerve hypoplasia should be evaluated for neuroendocrine deficiency and by MRI for midline brain malformation (138). A large number of genetic mutations have been demonstrated in Kallmann syndrome and hypogonadotropic hypogonadism (85; 243). Biallelic mutation in the gene EXOC3L2 causes a rare syndrome affecting the brain with visual impairment, pituitary hypoplasia with hormonal deficiencies, and kidneys and blood; the mechanism involves trafficking of post-Golgi vesicles to the plasma membrane (214). SEMA3A mutation at the 7q21 locus (a gene involved in hypothalamic neuroblast migration) causes short stature and multiple pituitary hormonal deficiencies that may not be recognized until adult life in some cases, and also can be associated with dysplasias of the heart, kidney, and long bones (101). An epigenetic phenomenon by prenatal alcohol exposure in mice copies the Cdon mutation by impeding Shh function and is another etiology of optic nerve hypoplasia and may be relevant to human fetal alcohol spectrum disorder (111).
Mitchell and colleagues stated that “HESX1 is the only gene associated with ectopic posterior pituitary lobe” (167) and other authors have confirmed this HESX1 mutation with ectopic neurohypophysis (238), in a large series of 83 patients (194), and in a familial pedigree with consanguineous parents; the phenotype is variable with the same HESX1 genotype, even amongst siblings in the same family (72). Ectopic neurohypophysis may be associated with extrapituitary cerebral malformations but is more frequently an isolated finding (32). In sum, it is now evident that no single gene can account for all of the anterior pituitary aplasias and dysplasias and the extreme variability in associated brain, eye, and somatic abnormalities, but posterior pituitary ectopia or dysplasias may be due to a single or more limited number of mutated genes.
Isolated growth hormone deficiency is usually due to GH1 and GHRHR mutations, but numerous other novel mutations have also been demonstrated in isolated cases associated with under stature (250). Similarly, there are many genes identified in cases of central congenital hypothyroidism, but they are different genes from those of growth hormone deficiency (212). Some antiepileptic medications may affect thyroid hormonal function as an epigenetic phenomenon (96), and some pregnant women who have epilepsy require medication during pregnancy and, thus, the fetus may be affected. Pituitary thyroid-stimulating hormone may remain normal in some.
Documentation states that mice lacking the homeobox gene Hesx1 exhibit variable anterior CNS defects (53). These comprise a reduced prosencephalon, abnormalities in the corpus callosum and septum pellucidum, anophthalmia or microphthalmia, defective olfactory development, and pituitary dysplasia. Septo-optic dysplasia is a comparable and highly variable phenotype in humans (54). HESX1 mutations were associated with isolated congenital pituitary hypoplasia and septo-optic dysplasia in 228 patients (238). The association of septo-optic-dysplasia with pituitary aplasia and ectopic neurohypophysis is so well documented that this malformation is now generally called “septo-optic-pituitary dysplasia,” though not all cases include pituitary defects (159; 213; 146; 240). Aplasia of the neurohypophysis has even been demonstrated at autopsy in a fetus of 23 weeks gestation (108). It also may be seen in some cases of holoprosencephaly (160). A novel homozygous HESX1 mutation, however, can cause isolated panhypopituitarism without midline defects or optic nerve hypoplasia (63).
Another important mutation associated with ectopic neurohypophysis, growth hormone deficiency, or panhypopituitarism is OTX2, often with a microdeletion at locus 14q22.3 and consisting of either frameshift or nonsense mutations (110; 148; 217). The endocrinopathy is often easy to suspect in this genetic defect because of the phenotype clinical markers in involved patients: anophthalmia or microphthalmia, at times agenesis of the left carotid artery, and intellectual deficits.
A small supernumerary marker chromosome resulting in a tetrasomy 22pter-22q11.21 is the characteristic genetic mutation in the cat eye syndrome, often with Mendelian transmission, hence, familial cases (107). Malformation of the hypothalamic-pituitary axis and growth hormone deficiency are demonstrated in affected children, as well as ocular iris configuration with a dorsoventrally elongated pupil resembling felines.
Quentien and colleagues found that “the transcription factor Pitx2 is required for the morphogenesis of anterior structures such as the eye, teeth, and anterior pituitary” (187). As the “Pit3 gene belongs to the family of RIEG/PITX genes…it is transcribed in the presumptive pituitary already at the open neural tube stage. During further development, Xpitx3 is strongly transcribed in the pituitary Anlage, the lens placodes and head mesenchyme, respectively” (183).
Pituitary dysfunction or hypoplasia may be associated with Fanconi anemia syndrome (112). In children with congenital melanocytic nevi, including neurocutaneous melanocytosis (a postzygotic mosaic RASopathy), the pituitary may be abnormal structurally and functionally, particularly with respect to growth hormone and insulin insensitivity (247).
Chromosomopathies associated with pituitary dysplasia and insufficiency include a 1q24.3q31.1 deletion in a girl who also had congenital lipomatosis of the face (40; 250), but chromosomal deletions, duplications, or translocations generally are rare in this disorder. Ring chromosome-18p deletion, however, results in anterior pituitary aplasia, with its resulting multiple neuroendocrinopathies (25). The male newborn of a mother with Turner syndrome (45X0) had severe panhypopituitarism and anatomical pituitary stalk interruption syndrome (ectopic neurohypophysis) and a normal 46XY karyotype; most women with Turner syndrome are infertile, but 2% to 8% can become pregnant (175).
Little is known about the influence of the genotype on the pituitary gland morphology and on the appearance of the sella turcica. The first systematic prenatal study is of the region in 14 human trisomy 18 fetuses (134). In all of the fetuses, pituitary dysplasia occurred with pharyngeally located adenopituitary tissue and malformation of the sella turcica. The pharyngeal tissue showed a positive reaction to immunohistochemical marking for thyroid-stimulating hormone. Postnatally, Taine and colleagues found pituitary dysplasia in a girl with an 18p deletion, total growth hormone deficiency, and a single central maxillary incisor (232). In a study on sella turcica pituitary gland development in prenatal trisomy-21, four different morphological types were described (135). The appearance of the most frequently occurring morphological type resembled normal appearance (type I).
To further elucidate the influence of the genotype on the phenotypic development in the pituitary gland region, prenatal and postnatal charting of the malformation fields in different genotypes, either histologically (162; 28; 130) or by CT or MRI scanning (mapping sella turcica morphology and the possible presence of a craniopharyngeal canal) (137), are important. Abnormal morphology of craniofacial tissues, including the adenohypophysis, has been described in a fetus with hypohidrotic ectodermal dysplasia (173). Animal studies generally correlate well with human data. The transcription factor of the insulin gene-enhancing protein isl2a is a regulatory factor in pituitary development and subsequent function in zebrafish (255).
Duplication of the pituitary stalk and hypophysis. This rare malformation is probably due to upregulation of a ventralizing gene. It can be associated with other midline cerebral defects, precocious puberty, and 2p12 deletion (184). It is also reported in association with nasopharyngeal teratoma, hypothalamic hamartoma, and cleft palate (44) or with benign nasal hamartoma (86). It is also described with pituitary adenoma and aqueductal stenosis, and with vertebral anomalies (180; 164). Association with heterozygous deletion of chromosome 14, in which the gene encoding thyroid transcription factor-1 is localized, is yet another different condition (02). ROBO1 deletion is associated with pituitary stalk interruption (ectopic neurohypophysis) but also duplication; this gene is known to be involved in neurogenesis and axonal guidance (211). In sum, the duplication of the pituitary stalk is probably multifactorial and due to multiple genetic defects, unlike ectopic neurohypophysis. A rare hereditary pituitary hyperplasia without stalk duplication is also described with infantile gigantism (88). Rathke cleft cysts are associated with pituitary dysplasia and a particular involvement of gonadotropic hormones, especially in pubertal girls who show estrogen receptor immunoreactivity in pituitary cells but not in the cells lining the cyst (115).
Ectopic neurohypophysis. This term refers to an interrupted or greatly thinned pituitary stalk with ventral displacement of the posterior pituitary. The diagnosis is based on MRI findings. Neuropathological examination to confirm the status of the pituitary stalk is rare because treatment is nonsurgical and autopsy findings are not well documented in this regard and are prone to artifactual disruption even if considered before brain removal. Gender and ethnic predispositions to ectopic neurohypophysis are not noted. An incomplete or partial form of ectopic posterior hypophysis is recognized by MRI (11). Other cerebral anomalies may coexist with ectopic neurohypophysis in some patients: optic nerve hypoplasia, cerebellar hypoplasia, and abnormal intracerebral vessels (06).
The first recorded description of ectopic neurohypophysis was a neuropathological study in 1927 by Priesel (185). The entity was then essentially forgotten for many decades and rediscovered in the era of MRI by Fujisawa and colleagues in a 1987 paper (78). An ectopic or ventrally displaced posterior pituitary gland was then reconfirmed on MRI by numerous subsequent authors (118; 121; 01; 154; 16; 39; 241; 181; 97; 139; 45; 167; 260; 259; 233; 244; 104; 155; 201; 258; 245). It is most readily recognized by the intense unenhanced T1-weighted signal of the neurohypophysis, normally seen on MRI at the median eminence in the floor of the 3rd ventricle, but it can also disclose an ectopic location. The increased T1 signal is attributed to neurosecretory vesicles, more specifically, an enhanced relaxation of water protons adjacent to these vesicles (35; 260). The T1 signal is enhanced by gadolinium.
More than 90% of publications on the topic appear in the radiology literature, with additional publications in genetics and endocrinology journals, but almost none are found in the neurologic literature. Multiple genetic mutations have been reported in both isolated cases and in ectopic neurohypophysis associated with other brain anomalies (06).
Neurologic manifestations of ectopic neurohypophysis. The clinical expression depends largely on the other associated cerebral malformations and may be epilepsy, cognitive and intellectual deficits, and motor deficits such as pseudobulbar palsy and spastic diparesis. Endocrinologic manifestations are usually an isolated growth hormone deficiency, and this may lead chronically to short stature or dwarfism if not recognized early and treated with exogenous growth hormone (241; 45). In adolescent or young adult girls, short stature and amenorrhea may be presenting symptoms (264). Multiple defects of the hypothalamic-pituitary axis with panhypopituitarism may also occur (45; 190). Abnormal ADH secretion and diabetes insipidus, as might be expected with posterior pituitary defects, are rare or absent. Neuroimaging demonstration of ectopic neurohypophysis can be correlated with the presence of growth hormone deficiency (58). Hypopituitarism associated with ectopic neurohypophysis may complicate some cases of static encephalopathies with spastic diplegia (ie, cerebral palsy); therefore, at least a limited endocrine panel should be included in the investigation of such children (242). Hypopituitarism discovered in young adulthood may prompt imaging to confirm an ectopic neurohypophysis (263). Ectopic neurohypophysis is associated in some cases with developmental brain malformations, including perisylvian polymicrogyria and cerebellar dysplasia (239). Joubert syndrome of cerebellar and brainstem anomalies (230), and also suprasellar arachnoidal cysts (209). Renal anomalies also are reported in some cases of ectopic neurohypophysis, so renal status should be assessed (177). Poland syndrome (absence of pectoralis major muscle) can be accompanied by ectopic neurohypophysis, associated with a 1.5Mb Xp22.31 duplication (158).
Though isolated ectopic neurohypophysis or pituitary stalk interruption syndrome is not generally associated with epilepsy, except in a minority of patients with septo-optic-pituitary dysplasia, an association with focal cortical dysplasia may occur in some, and epilepsy may be the presenting symptom in these cases (07).
Etiology and pathogenesis of ectopic neurohypophysis. The most common predisposing factor in the etiology of ectopic neurohypophysis is believed to be genetic, and familial cases are well documented (118; 01; 154; 16; 241; 181; 97; 139; 45; 194). Mutated genes in a minority of cases are HESX1 (238) and LHX4 (233). HESX1 is a gene implicated in septo-optic-pituitary dysplasia, a malformation in which ectopic neurohypophysis is a frequent component, and HESX1, in addition, is essential for pituitary development in the embryo. A novel nonsense mutation in the GLI2 gene has also been reported (76). Other genetic mutations associated with ectopic neurohypophysis include SOX3 (Xq26.3-27.3) (08; 225), GPR161 in a family identified by whole-exome sequencing (114), ROBO1 (23; 211), digenic PROKR2 and WDR 11 (161), a lethal autosomal recessive LHX4 (92), and in 18p deletion (256). It is also reported in Turner syndrome (45XO) (66). The ciliopathy TTC26, due to a homozygous mutation, can produce this pituitary phenotype (56). A genetic cause is, thus, likely. The original etiological explanation of traumatic transection of the pituitary stalk at birth, particularly with breech delivery, is no longer tenable. Many infants with ectopic neurohypophysis are born by vertex vaginal delivery or even by Cesarean section. Birth trauma also would not explain the association with other malformations of the brain.
Clinical vignette.
Ectopic adenohypophysis. Although ectopic neurohypophysis is common (see below), ectopic adenohypophysis is rare but is reported in the nasopharynx as a thyroid-stimulating hormone (TSH) secreting lesion (125).
The incidence of pituitary aplasia and dysplasia is unknown. It would be important in the future to specify the incidence of pituitary aplasia and dysplasia across the different types of malformation and the inheritance within each type (200; 29). Subsequent studies using modern neuroimaging suggest that the incidence is much higher than previously appreciated (178). The MRI findings that have led to this recognition not only are visualization of the pituitary itself but increased recognition of the extracerebral malformations in septo-optic-pituitary dysplasia that account for a majority of cases (138; 82; 186), and specific genetic mutations are now being recognized, such as FEVR and FLNA (74; 261). Anterior pituitary atrophy also can be associated with Rathke cleft cyst, craniopharyngioma, local pituitary fossa arachnoidal cysts, and other anatomical lesions (79; 254). Patients with pituitary stalk interruption syndrome (ectopic neurohypophysis) are divided into syndromic (associated with other extra-pituitary brain malformations; 48%) and nonsyndromic (isolated) forms, with the syndromic showing the most severe anatomical and hormonal alterations (22).
Geographical distributions are sometimes noted: there is an increased incidence of septo-optic-pituitary dysplasia in northern Canadian territories where the population is mainly indigenous and poverty, poor nutrition, and health issues are widespread; these exogenous conditions affect pregnant mothers and fetuses living in these regions (124). Another epidemiological focus worldwide is maternal age; in a large pan-European EUROCAT study, the prevalence of septo-optic-pituitary dysplasia was found to be between 1.9 and 2.5 per 100,000 live births, with the highest prevalence in those born to mothers aged 20 to 24 years (82). This prevalence in young mothers contrasts with most disorders in which older maternal age is a risk factor.
No known prevention for pituitary aplasia and dysplasia exists. Genetic considerations have been in focus in this context (48). If the administration of folic acid prevents nonclosure of the human neural tube (62), it is probable that folic acid can also prevent pituitary dysplasia. The risk connected with pituitary aplasia and dysplasia is the development of endocrinologic disorders, conditions that can be treated by hormone substitution. The extent to which pharyngeally located adenopituitary tissue develops into craniopharyngioma is not known. It is probable that, in many craniofacial malformations, pharyngeally located pituitary tissue is present but never develops into craniopharyngioma. Pharyngeally located adenopituitary tissue has, on prenatal autopsy material, shown expression of adenopituitary hormones (126). Whether this expression has clinical significance is not known.
In a 9-month-old male infant with sacral agenesis and caudal regression syndrome who also had growth hormone deficiency, endocrine replacement therapy had the additional beneficial effect of promoting distal muscle innervation of the lower extremities (61). A novel pathogenic variant in OFD1 results in X-linked Joubert syndrome, a ciliopathy, with orofaciodigital features and pituitary aplasia (12). Pituitary stalk interruption may occur in the TTC26 mutation, another ciliopathy (56).
Clinically, the postnatal diagnosis of pituitary aplasia and hypoplasia is based on endocrinological charting, blood-glucose measurement, radiography, and CT or MRI scanning of the sella turcica region. The postnatal and prenatal autopsy diagnosis is based mainly on histological examination of the brain and the sagittal cranial base block, including the pituitary gland.
Hormonal deficiencies involving hypofunction of the hypothalamus or the anterior or posterior portion of the pituitary gland may be found in conditions other than pituitary aplasia and dysplasia. Conditions where the target organ is malfunctioning (eg, myxedema) or the target receptors are defective (eg, Laron syndrome) (defective growth hormone receptors) mimic hypofunction of the specific pituitary hormone. These conditions, however, are usually limited to a single pituitary axis, making the distinction easier. Some target organ failures may further be distinguished from the corresponding pituitary function by specific signs and symptoms (eg, the hyperpigmentation in Addison disease) that are absent in hypopituitarism.
Neonatal hypoglycemia in pituitary aplasia and dysplasia may present with signs and symptoms similar to ketotic hypoglycemia (ie, severe hypoglycemia with ketonuria between meals). The conditions may be differentiated by the adrenocorticotropin, cortisol, and growth hormone deficiency present in the former. Furthermore, ketotic hypoglycemia tends to present later in life (18 months to 5 years).
For the purpose of CT or MRI differential diagnosis, it is important to be aware of the "empty sella" syndrome. Empty sella is not a disease but an anatomical state with characteristic radiological findings caused by many different pathological conditions requiring correct interpretation on each occasion (224). Empty sella is the radiological appearance of a sella turcica that is partially or completely filled with cerebrospinal fluid. Empty sella is common in adults, with a prevalence rate from autopsy of approximately 6% (49). The term "empty sella syndrome" should be used when an empty sella is present in association with headaches, visual disturbances, papilledema, or endocrine dysfunction. The precise etiology of empty sella is not known. Empty sella syndrome is rare in pediatric patients as compared to adults (266). However, approximately one-half of the children with growth hormone deficiency have a primary empty sella (231; 182). In a review of 175 adults with empty sella syndrome, endocrinopathy due to pituitary insufficiency was found in two thirds of cases, with a strong male predominance (93).
Rathke cleft cysts typically appear cystic with T1 hyperintensity and variable T2 signal and thin or no marginal enhancement, remaining stable over time (79). Persistence of the craniopharyngeal canal occurs with ectopic neurohypophysis secondary to SOX3 gene deletions in both the mouse and human (08). Persistent expression of activated Notch in the developing hypothalamus affects the survival of pituitary progenitor cells and alters pituitary structure (17). In rare instances, large pituitary colloidal cysts may occur (80). Epidermoid cysts of the isolated pituitary stalk have also been reported and can be successfully surgically resected (144).
McCune-Albright syndrome is a triad of polyostotic fibrous dysplasia of bone, precocious puberty, and cutaneous café-au-lait spots, but many cases later develop multiple endocrinopathies, particularly excessive growth hormone secretion (59; 257). Another bony dysplasia, Desbuquois dysplasia type 2, can be associated with growth hormone deficiency and XYLT1 mutations (106).
Ectopic prolactin-producing pituitary adenoma is reported rarely within a benign ovarian cyst teratoma (10). Congenital pituitary hypoplasia with hypopituitarism from birth may be associated with agenesis of the internal carotid artery (103).
Traumatic brain injury may follow closed head trauma in early childhood and may result in pituitary dysfunction and hormonal abnormalities, including precocious puberty, which should be treated and distinguished from congenital dysplasias of the pituitary or genetic defects affecting pituitary function (60; 223).
When pituitary aplasia and dysplasia is suspected, revealing pituitary dysfunction by measurements of pituitary and target organ hormone levels is a key procedure in establishing the diagnosis. Demonstration of abnormally low growth hormone levels is cardinal. Funduscopic examination should be performed by an ophthalmologist in all cases, not only for associated optic nerve hypoplasia but also for more subtle findings such as optic disc pits in ectopic neurohypophysis (100).
Growth hormone is released episodically, and random plasma levels in normal subjects are low, often at the level of detectability. A range of provocative tests that increase plasma growth hormone levels has been established. The drugs used to stimulate growth hormone release are L-dopa, clonidine, glucagon, insulin, or arginine. Peak growth hormone levels below 7 ng/mL during testing suggest growth hormone deficiency. Two or more of these tests must be positive in order to consider the patient to be growth hormone deficient.
Spontaneous growth hormone levels may be evaluated with a 24-hour test. During the 24-hour test period, blood is drawn every 20 minutes to 30 minutes, and the height and frequency of the plasma growth hormone peaks are evaluated. This type of procedure may enable diagnosis in children with abnormal pulsatile function of growth hormone release. However, in children with absent growth hormone secretion, this test is usually unnecessary in the diagnostic workup.
Plasma levels of insulin-like growth factor 1 reflect those of growth hormone and may be used to indicate growth hormone deficiency. In healthy children, levels of insulin-like growth factor 1 gradually increase from birth and peak at puberty. In growth hormone-deficient children, insulin-like growth factor 1 levels are initially low but rise within 16 to 28 hours of growth hormone administration.
The finding of hypofunction of the pituitary-thyroid axis in the neonatal thyroid screening programs may be the first sign of pituitary aplasia or dysplasia. Levels of several pituitary hormones, along with the target hormone plasma levels, should be determined. Thus, full workup includes measurement of plasma thyroid-stimulating hormone, liothyronine, thyroxine, corticotropin, cortisol, follicle-stimulating hormone, luteinizing hormone, sex steroids, prolactin, and antidiuretic hormone. Interpretation of these results allows for differentiation between target organ failure and pituitary or hypothalamic failure. However, these tests may not allow for a differentiation between pituitary and hypothalamic failure.
Congenital hypopituitarism could be due to hypothalamic failure (19); thus, tests that bypass the hypothalamus should be performed to establish the diagnosis of pituitary aplasia or dysplasia. One such test is the combined anterior pituitary test that involves simultaneous intravenous administration of four releasing hormones: (1) growth hormone-releasing hormone, (2) corticotropin-releasing hormone, (3) luteinizing hormone-releasing hormone, and (4) thyrotropin-releasing hormone. Plasma levels of the pituitary hormones are measured. In addition to the relevance of the combined anterior pituitary test in evaluating the target hormone plasma levels, it may be used to screen patients with suspected pituitary dysfunction. Central hypothyroidism is another expression, including in septo-optic-pituitary dysplasia, readily treatable with replacement exogenous hormone (26).
Pituitary aplasia and dysplasia may present as moderate to severe hypoglycemia with ketonuria with missed meals. Spontaneous blood glucose levels from 9 mg/dL (0.5 mM) and greater have been reported. Cortisol or growth hormone deficiency may cause hypoglycemia due to decreased gluconeogenic enzymes in cortisol deficiency, increased glucose utilization with lack of antagonistic effects of growth hormone on insulin action, or failure to supply endogenous gluconeogenic substrates with compensatory breakdown of fat and generation of ketones. In this manner, hypoglycemia in pituitary aplasia and dysplasia presents similarly to ketotic hypoglycemia.
Diagnostic imaging. MRI is essential to demonstrate pituitary anomalies and other brain malformations, including optic nerve hypoplasia and agenesis of the septum pellucidum (52; 170; 254; 186; 51; 43; 203). MRI is a good predictor of the severity and type of growth hormone deficiency in children (215). Associated disorders of neuroblast migration and the absence of olfactory bulbs, tracts, and sulci in 8.6% of cases are also revealed (203). Ectopic neurohypophysis (pituitary stalk interruption) and other anatomical pituitary anomalies are demonstrated in understature children with isolated growth hormone deficiency; enlargement (hyperplasia) of the pituitary may be seen in hypothyroidism (254). In a study of 50 children with precocious puberty, MRI revealed pathological findings often involving the pituitary in 17% of girls and 55.5% of boys (122). However, caution must be taken in interpretation because some children and adolescents exhibit pituitary defects by CT or MRI that are incidental findings and are not associated with hormonal aberrations (71; 143). Pituitary aberrations are also occasionally demonstrated as incidental, unanticipated findings in children without endocrinopathies or major brain malformations and may have an incidence as high as 10% of normal subjects (178). Rapid MRI protocols not only recognize ectopic neurohypophysis but also demonstrate other midline structural abnormalities in the brain (151).
High-resolution MRI and newer MRI techniques, such as MR elastography, perfusion imaging, diffusion-weighted and diffusion-tensor imaging, heavily weighted T2, and spectroscopy, can now provide even more diagnostic information about the abnormal pituitary in children (33; 70). A large, international database demonstrates the correlation of pituitary anomalies with clinical endocrinological deficiencies and association with optic nerve and other structural abnormalities of the brain (58). The diagnosis of pituitary aplasia and dysplasia should not be made without appropriate attempts to image the sella turcica region. MRI of the pituitary and suprasellar region has become much more refined and precise in recent years (99) and even extends to prenatal fetal MRI (41; 195). Although plain skull x-rays may be employed, a more precise examination may be performed with high-resolution CT or, whenever possible, MRI. When the scans reveal a low-density area in the intrasellar region consistent with cerebrospinal fluid, the empty sella diagnosis may be relevant. The diagnosis of an empty sella is made in 20% to 50% of children with pituitary dysfunction (182; 05). A shallow sella turcica may be seen in association with a normal pituitary gland (226). Although the imaging techniques may reveal low-density areas in the sella region, they generally fail to reveal pituitary ectopia. In many cases, however, a small anterior pituitary lobe and lack of visualization of the pituitary stalk may be seen on MRI (234). Pituitary aplasia, dysplasia, and ectopia were found in a considerable number of cases in several postmortem investigations of pituitary morphology in children who died from hypoglycemia secondary to hypopituitarism (226). It was demonstrated that MRI in pituitary dwarfs can reveal unusual intrasellar findings such as pituitary aplasia (267). Familiar hypopituitarism represents a clinically and genetically heterogeneous disorder. In a subset of these families, defects in Pit-I, a transcription factor essential for proper pituitary development, have been identified as an underlying molecular cause. These patients present with extreme short stature as well as growth hormone, prolactin, and thyroid-stimulating hormone deficiency; however, patients present with intact adrenocorticotropic hormone, luteinizing hormone, and follicle-stimulating hormone secretion. MRI evaluation of the pituitary indicated pituitary aplasia in all subjects (172).
The key to successful management is hormone replacement therapy administered based on clinical and laboratory findings, including skeletal maturity measures. The replacement therapy rests on the administration of the hormones produced by the target glands, except in cases of growth hormone and antidiuretic hormone deficiency, when growth hormone and desmopressin acetate, respectively, are administered.
The recommended growth hormone substitution dose is 0.15 to 0.30 mg/kg per week, administered subcutaneously in three divided doses to seven divided doses. The medication may be administered in the clinic or at home by the parents. Compliance with the latter has improved with pen injection systems. The maximal response to the treatment is expected in the first years. Therapy should be continued at least until closure of the epiphyses and clinically determined growth arrest.
Thyroid substitution in the form of sodium-L-thyroxin administered orally is the treatment of choice. In neonates, the dose is 10 to 15 µg/kg per day, reduced to 4 µg/kg per day in children, and 2 µg/kg per day in adults. Monodeiodination of thyroxine to liothyronine is the basis for the restoration of normal plasma levels of liothyronine.
Glucocorticosteroids should be substituted at doses not exceeding the equivalent of 15 mg/m2 per day of hydrocortisone, preferably as intermediate-acting drugs such as prednisone. The hormones are administered twice daily. A larger dose is given in the morning, and the remainder is given before dinner to mimic the physiological circadian hormone release. It should be stressed that a two- to fourfold increase in glucocorticosteroid administration may be needed initially in stressful situations that increase glucocorticoid requirements. Otherwise, the patient may go into a severe adrenal crisis. Administration of corticosteroids with mineralocorticoid effects in patients with pituitary aplasia and dysplasia is usually not necessary because adrenal release of aldosterone is maintained independently of adrenocorticotropin stimulation.
In boys with microphallus, a 3-month course of monthly intramuscular injections of 25 mg of testosterone enanthate may enlarge the penis without effects on bone maturation. Sex steroids or gonadotropins may be indicated at the time of normal puberty onset or later, determined by skeletal maturity.
Treatment of posterior pituitary hypofunction is less essential. Oxytocin replacement therapy is generally not appropriate. Antidiuretic hormone deficiency leads to diabetes insipidus, a life-threatening condition in infants who cannot consciously regulate their liquid intake, but it is less of an issue in adults. The treatment of choice is desmopressin acetate, a long-acting synthetic nonapeptide analog of antidiuretic hormone. The dosage for symptomatic relief in adults is 10 to 40 µg daily in two or three divided doses, either as tablets or as a solution inhaled deeply through the nose. In children, 5 to 10 µg of desmopressin acetate daily may be given.
If ectopic neurohypophysis is associated with other brain malformations, the risk of epilepsy is increased and should be treated with antiseizure medications. Sudden death is even reported in rare cases of Joubert syndrome with ectopic neurohypophysis (230).
Endocrine testing should be done periodically in children with growth hormone deficiency because over time they may also develop other pituitary hormonal insufficiencies that were not present earlier (31).
Treatment of these patients requires close monitoring of physical and psychological development as well as repeated lab tests. The question of gene therapy has been put forward. Thus, Davis and McNeilly concluded that “gene therapy for pituitary disease is likely to be feasible in the future but will require careful and extensive evaluation of efficacy and safety, using a variety of possible methods of gene delivery” (57).
Pituitary aplasia and dysplasia is a congenital condition that may be treated with substitution therapy. The condition, however, is permanent, and normal pituitary function cannot be obtained. The mortality varies and is closely related to the underlying lethal condition (ie, anencephaly or severe cases of holoprosencephaly). When the disorder is found in normocephalic children and appropriate substitution therapy is instituted, the prognosis quo ad vitam is substantially improved.
The complications of pituitary aplasia and dysplasia may be a result of the condition itself or a result of the adverse effects of the treatment. When the condition is treated appropriately, the complications are significantly reduced. Lack of gonadotropins may delay pubertal development. In spite of appropriate hormone substitution therapy, short stature and abnormal body proportions are found. Progressive worsening in endocrinopathy throughout childhood may occur, so periodic review of hormonal status is needed, even in children on replacement therapy (22).
The adverse effects are mostly reversible and will not be dealt with here. A few adverse effects do, however, deserve special attention. Patients treated with growth hormone have a risk for leukemia that may be double the risk found in the general population (24). Patients treated before the era of recombinant growth hormone are at risk for developing Creutzfeldt-Jakob disease. Growth hormone and glucocorticoids are diabetogenic.
A 28-year-old Chinese woman developed pituitary stalk interruption demonstrated by MRI with multiple endocrinopathies following in vitro fertilization and embryo transfer (262). She was treated with hormonal replacement therapy and delivered by Cesarean section a healthy male neonate who is developmentally and endocrinologically normal at 1 year of age.
It is recommended that adult glucocorticoid-deficient patients be given a stress dose of 100 mg of hydrocortisone intravenously prior to surgery and repeated every 12 to 24 hours for a few days postoperatively (165).
Preferably, patients should be euthyroid prior to surgery by administration of thyroid replacement therapy. Body heat mechanisms are inadequate in hypothyroid patients, and the temperature should be monitored and maintained.
Patients with inadequate antidiuretic hormone secretion are given a preoperative bolus of 0.4 µg desmopressin acetate intranasally or intravenously, followed by a constant infusion of 0.4 µg/hour to 0.8 µg/hour. Hypertonic fluids should be avoided intraoperatively, and plasma and urine osmolality should be monitored (117).
Deficiency of prolactin, gonadotropin, and growth hormone usually does not require perioperative precautions. Special attention should be paid to blood glucose and cardiac function. Death can result during anesthesia for MRI or for surgery, including dental procedures in children (193).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Harvey B Sarnat MD FRCPC MS
Dr. Sarnat of the University of Calgary has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Developmental Malformations
May. 08, 2026
Developmental Malformations
May. 08, 2026
Developmental Malformations
May. 08, 2026
Developmental Malformations
May. 08, 2026
Developmental Malformations
Apr. 24, 2026
Developmental Malformations
Apr. 24, 2026
Developmental Malformations
Apr. 24, 2026
Developmental Malformations
Apr. 16, 2026