



















































Neurobehavioral & Cognitive Disorders
Mental status examination
Jun. 17, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Vitamin B12 deficiency may cause an extraordinary variety of progressive neurologic syndromes. In this article, the author discusses the manifestations of vitamin B12 deficiency.
|
• Vitamin B12 deficiency should be suspected in any patient with otherwise unexplained peripheral neuropathy, myelopathy, optic neuropathy, dementia, ataxia, movement disorder, or psychiatric disturbance and in individuals with macrocytosis with or without anemia. | |
|
• Serum B12 level should be determined in any patient with suspected vitamin B12 deficiency. | |
|
• Abnormal red blood cell indices are neither sensitive nor specific for vitamin B12 deficiency. | |
|
• In cases of borderline low vitamin B12 levels, or when vitamin B12 deficiency is strongly suspected despite reported normal levels from an automated assay, elevated serum methylmalonic acid and homocysteine levels may confirm a physiological deficiency, as may a normalization of these levels with B12 repletion. | |
|
• Daily, high-dose oral vitamin B12 supplementation appears as effective as parenteral therapy and is substantially less costly. This has really only been tested, though, in minimally symptomatic or asymptomatic patients. A brief parenteral course of therapy may still be needed for patients with significant neurologic signs of vitamin B12 deficiency. |
At a meeting of the South London Medical Society in 1849, and subsequently, in a monograph in 1855, British physician Thomas Addison (1793–1860) at Guy’s Hospital in London described several cases with “idiopathic” anemia characterized by pallor, weakness, and progressively worsening health leading to death (139). Later, this condition was called Addisonian anemia, at least until German internist Michael Anton Biermer (1827-1892) in Zurich named it perniciöse Anämie (ie, pernicious or fatal anemia) when describing 15 cases of severe anemia (of mixed etiologies) in 1872 (139).
Although pernicious anemia, or Addison-Biermer disease, had been recognized clinically in the mid-19th century, the associated neurologic, gastrointestinal, and hematologic manifestations were not recognized clinically and linked with pernicious anemia until the end of the 19th century (139).
In 1884, Lichtenstein described cases of pernicious anemia with neurologic manifestations felt to be suggestive of tabes dorsalis (139). The first accurate description of spinal cord pathology associated with certain types of anemia was by German physician Ludwig Lichtheim (1845-1926), who described three cases, two with autopsy (149). Similar cases were reported by a number of authors over the next several decades (139). Guyanese-British neurologist James Samuel Risien Russell (1863-1939) and colleagues coined the term "subacute combined degeneration of the spinal cord" in their study of the neuropathological abnormalities commonly associated with pernicious anemia (217).
In 1870, British gastroenterologist Samuel Fenwick (1821-1902) in London associated stomach atrophy with this form of anemia and demonstrated that stomach mucosa from an affected fresh cadaver could not digest boiled egg white with prolonged incubation, whereas mucosa from a control stomach could do this (139). Subsequently, German internist Arnold Cahn (1858-1927) and German physician and physiologist Josef Freiher von Mering (1849-1908), working in the Strassburg clinic of German physician Adolf Kubmaul (1822-1902), showed that a patient with pernicious anemia had no hydrochloric acid in the stomach contents, a finding later demonstrated to be pervasive in this disorder and to precede the development of anemia (139).
It was not until the 1850s--after Addison’s original communication--that the first red cell counts were done by German physiologist Karl von Vierordt (1818-1884) and that hemoglobin was discovered by German physiologist Otto Funke (1828-1879) (139). In 1875, American physician and educator William Pepper Jr. (1843-1898), then a lecturer in clinical medicine at the University of Pennsylvania in Philadelphia (and subsequently Professor of Clinical Medicine, Professor of the Theory and Practice of Medicine, and the longtime Provost of the university), noted the extreme hyperplasia of the bone marrow in patients with pernicious anemia (139). In 1880, German physician-scientist (and later Nobel laureate) Paul Ehrlich (1854-1915), using aniline dyes developed by his cousin Carl Weigert (1845-1904), a German pathologist, identified large erythroid precursor cells that he called “megaloblasts” in stained blood smears of patients with pernicious anemia (139). Subsequent hematologists noted characteristics of megaloblastic anemia in the peripheral blood (ie, macrocytes, poikilocytes, and hypersegmented neutrophils) and bone marrow (eg, megaloblasts, meta-myelocytes, and megakaryocytes) (139). Later, American physician-scientist Francis Weld Peabody (1881-1927) of the Thorndike Memorial Laboratory in Boston hypothesized that this macrocytic anemia was due to maturational arrest of erythroblasts in the bone marrow (139).
In 1925, American medical researcher George Minot (1885-1950), at Peter Bent Brigham Hospital in Boston, and American physician William Parry Murphy (1892-1987) at the Collis P Huntington Memorial Hospital of Harvard University, hospitalized a group of patients with pernicious anemia to systematically assess liver as a treatment (139). By 1926, Minot and Murphy reported clinical and hematological improvement in 45 patients with pernicious anemia treated with a dietary regimen that incorporated large quantities of liver (Minot and Murphy 1926; 139). The patients improved clinically, often dramatically so, in conjunction with improvements in their hematological indices. This suggested that a factor present in calf liver could rapidly restore red blood cell counts in pernicious anemia. Moreover, this clinical improvement could be sustained for many years, well beyond the previous life expectancy of such patients. Patients with relatively mild neurologic dysfunction also improved, but patients with more severe neurologic dysfunction showed, at best, slow and limited improvement. Eventually, with Edwin J Cohn, a physical chemist in the Laboratories of Physiology at Harvard Medical School, they tried to isolate the active principle in the liver, which resulted in clinical improvement. Although they were not successful in isolating the responsible factor in the liver, they did demonstrate that potent extracts could be given parenterally in very small quantities. In 1934, Minot and Murphy were awarded the Nobel Prize for their discovery of a treatment for pernicious anemia.
In 1926, after Minot and Murphy’s success with liver therapy for pernicious anemia, William Bosworth Castle (1897-1990), then an assistant resident at the Thorndike Memorial Laboratory of Boston City Hospital (which had recently come under the direction of Minot as successor to Francis Peabody), decided to pursue his belief that gastric achlorhydria (“achylia gastrica”) was etiologically linked to pernicious anemia (139). Castle noted: (1) gastric achlorhydria precedes the other clinical manifestations of pernicious anemia, and (2) even when the blood of a patient with pernicious anemia is returned to normal with liver feeding, gastric achlorhydria persisted (139). Castle suggested that some essential step of gastric digestion was impaired, thereby disrupting the absorption of an essential dietary factor. He reasoned that this defective process might be circumvented by utilizing gastric juices from individuals with normal stomachs. By an ingenious series of experiments, Castle and his colleagues showed that indeed the normal stomach secretes a substance separate from normal gastric juice that is able to interact with a dietary substance to promptly relieve the anemia of these patients. Castle later labeled the essential substance secreted by a normal stomach as “intrinsic factor” and the substance present in food as “extrinsic factor.” Subsequent studies showed that the intrinsic factor serves as an intestinal transport vehicle for the extrinsic factor (later identified as vitamin B12) (139).
Cyanocobalamin (vitamin B12) was finally isolated by the mid-20th century, and this greatly improved the treatment of pernicious anemia and the associated neurologic manifestations (139). By 1955, British chemist Dorothy Crowfoot Hodgkin (1910-1994) of Cambridge University determined the molecular structure of cyanocobalamin using computer-assisted x-ray crystallography, work for which she received the 1964 Nobel Prize in Chemistry (139). The complex structure of vitamin B12 included a single cobalt atom at the center of a tetrapyrrole or “corrin” macro-ring structure. A complete chemical synthesis of vitamin B12 was finally achieved in 1960 by an international consortium of chemists.
Subsequent biochemical work demonstrated that only two enzyme systems require forms of vitamin B12 in man: adenosylcobalamin in the conversion of methylmalonyl coenzyme A to succinyl coenzyme A by methylmalonyl-coenzyme A mutase, and methylcobalamin in the conversion of homocysteine to methionine by methionine synthase (139).
In the late 1950s, studies showed that there are two mechanisms involved in vitamin B12 absorption. With physiologic (ie, 1 to 2 mg) doses of oral vitamin B12, the vitamin is absorbed by a process dependent on intrinsic factor; but with much larger oral doses, vitamin B12 appears in plasma much sooner as a result of passive diffusion, independent of the presence or absence of intrinsic factor. When vitamin B12 is released from foods by peptic digestion, it is bound to intrinsic factor, affording partial protection against gut microorganisms and parasites during transport through the gut to the terminal ileum, where the complex binds to microvilli of the intestinal epithelial cells.
Beginning in the late 1950s and continuing through the 1960s, several lines of evidence converged in support of an autoimmune basis for pernicious anemia: (1) corticosteroids improve vitamin B12 absorption and reduce anemia; (2) gastric and serum autoantibodies to intrinsic factor and gastric parietal cells are present in the majority of patients; and (3) other autoimmune diseases (eg, Hashimoto thyroiditis, insulin-dependent diabetes mellitus, Addison disease, and vitiligo) are common in such patients (139).
Pernicious anemia is now understood to be fundamentally an autoimmune disorder that begins with an autoimmune gastritis in which antiparietal cell antibodies produce atrophic gastritis with a resultant decline in intrinsic factor production, evolving over years or even decades (139). In 1988, the principal target of these antibodies was identified by Swedish internist F Anders Karlsson and colleagues as the acid-producing H+/ K+-adenosine triphosphatase (ATPase) in the cell membrane of gastric parietal cells (125).
The 2024 U.K. National Institute for Health and Care Excellence (NICE) guidelines on B12 deficiency proposed replacing the term "pernicious anaemia" with "autoimmune gastritis," a suggestion that has been appropriately criticized (250). Pernicious anemia develops in the advanced stages of autoimmune gastritis and is not synonymous with it (250). Pernicious anemia and autoimmune gastritis are different stages, and indeed different aspects, of the same disease continuum.
|
• Vitamin B12 deficiency typically presents with either hematologic or neurologic signs. | |
|
• The hematologic and neurologic signs associated with vitamin B12 deficiency may be dissociated. | |
|
• Well-recognized neurologic manifestations of vitamin B12 deficiency include myelopathy, peripheral polyneuropathy, optic neuropathy, psychiatric disturbances, and dementia. | |
|
• Subacute combined degeneration of the spinal cord refers to progressive degeneration of both the corticospinal (lateral column) and dorsal column tracts of the spinal cord. | |
|
• When present, the hematologic manifestations of vitamin B12 deficiency include macrocytic anemia, hyper-segmentation of neutrophil nuclei, neutropenia, and megaloblastic changes in the bone marrow. | |
|
• Hematologic abnormalities in the peripheral blood are insensitive indicators of even severe and neurologically symptomatic vitamin B12 deficiency. |
Vitamin B12 deficiency typically presents with either hematologic or neurologic signs. A curious feature of the condition is that the hematologic and neurologic signs may be dissociated. In fact, in one large series, the severity of the neurologic deficits correlated inversely with the degree of anemia and macrocytosis (96). There may also be cutaneous manifestations of vitamin B12 deficiency and additional manifestations associated with the underlying causes of the vitamin B12 deficiency.
Overall, the most common symptoms of pernicious anemia are fatigue (55%), loss of sensation in limbs (32%), excessive weight loss (27%), and a sore tongue (23%) (221).
Neurologic manifestations. Neurologic deficits are common in vitamin B12 deficiency. In one large study, 39% of patients with deficiency had neurologic manifestations, and in almost 80% of these cases, the neurologic symptoms were the sole or dominant manifestation of the deficiency (96). Well-recognized neurologic manifestations include myelopathy, peripheral polyneuropathy, optic neuropathy, psychiatric disturbances, and dementia.
Subacute combined degeneration of the spinal cord. This syndrome is so named because it may cause progressive degeneration of both the corticospinal (lateral column) and dorsal column tracts of the spinal cord. Typically, the neurologic syndrome evolves over a period of several months, although the disease may take a more chronic course. The most common early sign is paresthesia, which usually occurs first in the distal lower extremities. At this early stage, there may be no objective abnormalities on the neurologic examination. Later, signs and symptoms of a myelopathy involving the dorsal and lateral columns combined with a peripheral neuropathy may develop, but overt spasticity is uncommon (96). Initial ambulatory function is a useful clinical marker of the severity of spinal cord dysfunction and may be a useful predictor of final functional outcome (113). Impaired vibration and joint position sense are found on examination. The patient may be ataxic. Weakness is less common. Vitamin B12 deficiency only infrequently results in incontinence of bladder or bowel, impotence, or orthostatic hypotension.
Peripheral polyneuropathy. Vitamin B12 deficiency is a frequently recognized cause of distal symmetric axonal polyneuropathy, and it should be screened for in all patients presenting with polyneuropathy. Although commonly encountered in primary care, vitamin B12 deficiency may also be identified in patients previously deemed as having an idiopathic neuropathy seeking evaluation in a tertiary care center (74).
Uncommon neurologic manifestations. Rarely, vitamin B12 deficiency will manifest as a movement disorder, such as parkinsonism, focal dystonia, chorea, or blepharospasm (189; 04; 70; 226; 188). Rarer still are reports of seizures in vitamin B12 deficiency (65; 174; 177). Several small studies suggest that dysautonomic syndromes, such as orthostatic hypotension, postural orthostatic tachycardia syndrome (POTS), and syncope are associated with vitamin B12 deficiency and may be improved by vitamin B12 therapy (33; 171; 185; 78); however, dramatic dysautonomia does not appear to be a prominent manifestation of deficiency. Vitamin B12 deficiency may contribute to hearing impairment at low frequencies (0.25 to 4 kHz) in children, whereas pure-tone hearing thresholds from 8 to 16 kHz do not seem to be affected (09).
Infantile vitamin B12 deficiency. In Norway, where 5% to 10% of neonates and infants have biomarkers suggesting vitamin B12 deficiency from newborn screening tests and unselected clinical screening, spells, tremor, and irritability are common findings in early infant vitamin B12 deficiency (156). Nitrous oxide given during labor may be a contributing risk factor (156).
In a prospective study of 252 Norwegian infants aged 3 to 7 months, 46% had hyperhomocysteinemia, and 10% had hyperhomocysteinemia combined with clinically relevant symptoms suggestive of B12 deficiency (155); hyperhomocysteinemia was associated with tremor and excessive sleep, even though most infants with hyperhomocysteinemia did not show symptoms.
Severe Infantile vitamin B12 deficiency significantly impairs head growth, overall development, and body composition (263); it may also augment long-term cardiovascular risks due to abnormal lipid profiles.
Ophthalmologic manifestations. An unusual but well-documented manifestation of cobalamin deficiency is optic neuropathy. This may present as a subacute progressive decrease in visual acuity with a cecocentral scotoma (ie, a scotoma obscuring central vision and enlarging the blind spot). Vitamin B12 deficiency may present with a maculopathy similar to age-related macular degeneration, and it has been suggested that a relative deficiency may also play a role in age-related macular degeneration (AMD), the most common cause of blindness or low vision in older adults (64).
Dementia and neuropsychiatric manifestations. Severe vitamin B12 deficiency causes psychiatric and cognitive disturbances in some patients. The abnormalities are not specific and can range from depression or mild memory impairment to global dementia. Usually, these occur along with other neurologic deficits such as a myelopathy or neuropathy. It is uncertain whether mild or moderate vitamin B12 deficiency can cause dementia.
Uncommonly, vitamin B12 deficiency will present with prominent psychiatric manifestations. Psychosis has been reported several times (65).
Neurologic outcomes in infants and children. Cobalamin deficiency may occur in infants whose mothers are cobalamin-deficient or in those with rare inherited conditions such as Imerslund-Grasbeck syndrome (see Etiology section). Maternal vitamin B12 deficiency, exclusive breastfeeding, and a maternal diet low in vitamin B12 are associated with infant vitamin B12 deficiency, methylmalonic acidemia, and homocysteinemia (273).
Severe cobalamin deficiency in infancy presents as developmental regression. A severe cobalamin-deficiency neuropathy may result in "floppy infant" syndrome (210). Infantile spasms and West syndrome (developmental delay, infantile spasms, and hypsarrhythmia) have been reported to be potential outcomes of infantile dietary vitamin B12 deficiency (72; 159; 225). Myoclonus, tremor, or seizures may occur on initiation of vitamin B12 therapy in severely deficient infants; these conditions improve with continued vitamin B12 treatment (86; 87; 187; 36; 186). Infants and children who have experienced neurologic manifestations of vitamin B12 deficiency may be left with long-term neurologic impairment (265; 283; 246).
Non-neurologic manifestations. When present, the hematologic manifestations of vitamin B12 deficiency include macrocytic anemia with macro-ovalocytes and macro-erythrocytes (or macrocytes), hypersegmentation of neutrophil nuclei, variable polychromasia, neutropenia, and megaloblastic changes in the bone marrow. However, hematologic abnormalities in the peripheral blood are insensitive indicators of even severe and neurologically symptomatic vitamin B12 deficiency (96; 20). Severe vitamin B12 deficiency may cause a thrombotic microangiopathy resembling thrombotic thrombocytopenic purpura (183).
Gastrointestinal disorders that can be associated with vitamin B12 deficiency include atrophic gastritis, particularly in patients with pernicious anemia. Upper gastrointestinal endoscopy in atrophic gastritis shows a shiny surface and pale gastric mucosa with effacement of the gastric rugal (mucosal) folds, along with prominent submucosal vessels (206). Histopathological examination of biopsy tissue from the body and fundus of a patient with pernicious anemia may show areas of atrophy and a reduction in the number of glands, which are replaced by sheets of chronic inflammatory cells and fibrosis (206). In severe cases, a pan-enteropathy is present with diarrhea and malabsorption of nutrients. Rarely, jaundice occurs due to impaired erythropoiesis (62).
Cutaneous manifestations of cobalamin deficiency include most commonly hyperpigmentation but also hair and nail changes; oral changes (eg, atrophic glossitis or a "beefy red" tongue), and vitiligo (typically in association with pernicious anemia or autoimmune atrophic gastritis, specifically) (170; 34; 243; 258; 124; 220; 224; 223; 44; 191; 206; 256; 277; 54; 278; 31; 227).
(Source:Xu X, Liu Y, Xiong X, et al. Diagnostic value of oral "beefy red" patch combined with fingertip blood mean corpuscular volume in vitamin B12 deficiency. BMC Oral Health 2022;22[1]:273. Creative Commons Attribution 4.0 I...
(Source:Xu X, Liu Y, Xiong X, et al. Diagnostic value of oral "beefy red" patch combined with fingertip blood mean corpuscular volume in vitamin B12 deficiency. BMC Oral Health 2022;22[1]:273. Creative Commons Attribution 4.0 I...
(Source:Xu X, Liu Y, Xiong X, et al. Diagnostic value of oral "beefy red" patch combined with fingertip blood mean corpuscular volume in vitamin B12 deficiency. BMC Oral Health 2022;22[1]:273. Creative Commons Attribution 4.0 I...
(Source:Xu X, Liu Y, Xiong X, et al. Diagnostic value of oral "beefy red" patch combined with fingertip blood mean corpuscular volume in vitamin B12 deficiency. BMC Oral Health 2022;22[1]:273. Creative Commons Attribution 4.0 I...
Skin hyperpigmentation is typically seen on the hands and feet, but it can occur anywhere (162).
A 52-year-old Indian woman with megaloblastic anemia and peculiar cutaneous hyperpigmentation (prior to therapy). Photo shows diffuse, brownish-black discoloration of the palms (A1) and knuckle pad hyperpigmentation in the dors...
Resolution of hyperpigmentation on the palms (B1) and dorsum of the hands (B2) 12 weeks after initiation of parenteral cyanocobalamin therapy. (Source: Padhi S, Sarangi R, Ramdas A, et al. Cutaneous hyperpigmentation in megalob...
Hyperpigmentation and glossitis may resolve with vitamin B12 replacement (191).
Left untreated, pernicious anemia and other causes of severe vitamin B12 deficiency are fatal. Fatal cases are marked by severe myelopathy, encephalopathy, or anemia. Neurologic manifestations may not reverse with vitamin B12 therapy, so prompt recognition and treatment are needed to avoid permanent disability.
Pernicious anemia is also associated with an increased risk of carcinoid tumors and carcinoma of the stomach (236; 111). Endoscopic screening for gastric cancer may be warranted in patients diagnosed with pernicious anemia (15).
A 41-year-old woman was referred for headaches and numbness in the extremities. She first noted tingling paresthesias approximately 9 months earlier, beginning simultaneously in the feet and hands. Her past history was notable for migraine headaches and hypothyroidism; she had been on thyroid replacement therapy for 16 years. Her only prescription medication was thyroxin 125 µg daily. On examination, she appeared depressed and anxious. Cranial nerve and motor examinations were normal. There was mild difficulty with tandem walking, but the examiner perceived possible embellishment. Tendon reflexes were normal. Vibration sense was reduced in the great toes bilaterally, but sensory examination was otherwise normal. The Romberg sign was absent. Laboratory results included a normal thyroid-stimulating hormone level, normal hemoglobin A1c, and negative syphilis serology. Hematocrit and mean corpuscular volume were normal. A vitamin B12 level was 150 pg/ml (radioimmunoassay). Methylmalonic acid was elevated at 3500 nM/L. The patient was given cyanocobalamin 1 mg intramuscularly daily for 5 days (as an outpatient) and then maintained on 1 mg intramuscularly monthly. Her paresthesias and mild gait instability resolved within a month of initiating therapy. Vibration sense returned in the toes.
|
• Cobalamin participates as a cofactor in only two enzymatic reactions in humans: (1) conversion of homocysteine to methionine; (2) conversion of methylmalonyl-CoA to succinyl-CoA. | |
|
• Humans are completely dependent on dietary sources for cobalamin. | |
|
• Virtually all dietary cobalamin comes from meat or dairy products. | |
|
• Cobalamin is transported through the gastrointestinal system by carrier proteins (haptocorrin and intrinsic factor) until it is eventually taken up by receptor-mediated endocytosis in the terminal ileum. | |
|
• The carrier protein called "intrinsic factor" is a glycoprotein produced by gastric parietal cells. Intrinsic factor is necessary for receptor-mediated endocytosis in the terminal ileum. | |
|
• In the blood, cobalamin is also transported by carrier proteins, with the physiologically important fraction bound to the carrier protein transcobalamin II. | |
|
• Disturbances at any step of cobalamin metabolism may result in deficiency, but pernicious anemia accounts for greater than 90% of cases of symptomatic vitamin B12 deficiency, and most other symptomatic cases are due to other intrinsic factor-related sources of vitamin B12 malabsorption. | |
|
• Pernicious anemia is the result of an autoimmune gastritis in which the principal antigenic target is the H/K ATPase of gastric parietal cells. | |
|
• A deficiency in vitamin B12 is the most common nutritional deficiency in patients who have had gastric bypass surgery. Gastrectomy increases the risk of deficiency because parietal cells, the source of intrinsic factor, are removed. | |
|
• Pathology of the distal ileum (eg, from regional enteritis, Whipple disease, ileal tuberculosis, tropical sprue, and surgical resection of the distal ileum), where vitamin B12 is normally absorbed, may impair absorption of the intrinsic factor-cobalamin complex. | |
|
• Cobalamin deficiency is common in vegans, occurring in up to half of these individuals who do not receive vitamin B12 supplementation. | |
|
• Because nitrous oxide irreversibly oxidizes the cobalt in cobalamin, a functional deficiency of cobalamin can be caused by inhalation of nitrous oxide, either as an anesthetic agent or as a recreational drug. |
Cobalamin. Cobalamin is a complex molecule at the core of which is a corrin ring: a tetrapyrrole structurally homologous to heme but with a cobalt atom, rather than iron, at its center.
Cobalamin is known to participate in only two enzymatic reactions in humans (152).
Legend: AdoCbl, adenosylcobalamin; Cys, cystine; Cyst, cystathionine; Hcy, homocysteine; MeCbl, methyladenosylcobalamin; Met cycle, methionine cycle; MCM, methylmalonyl-CoA mutase; MMA, methylmalonic acid; MS, methionine syntha...
First, it is an essential cofactor for the conversion of homocysteine to methionine by the enzyme methionine synthase (MTR), or more precisely, the regeneration of methionine from homocysteine. Second, cobalamin is an essential cofactor for the conversion of methylmalonyl-CoA to succinyl-CoA by the enzyme methylmalonyl-CoA mutase (MUT). Cobalamin is not known to participate in DNA synthesis, but folate, as 5,10-methylene tetrahydrofolate, is essential for the synthesis of purines.
In the cytoplasm, vitamin B12 deficiency impacts the enzyme methionine synthase, with some consequences that adversely affect DNA, RNA, proteins, neurotransmitters, and phospholipids, and with some consequences that indirectly contribute to oxidative stress.
In mitochondria, vitamin B12 deficiency impacts the enzyme L-methylmalonyl-CoA mutase (MUT) with some consequences that adversely affect lipogenesis, as well as histone and DNA methylations processes.
Humans are completely dependent on dietary sources for cobalamin, with a minimum daily requirement of about 2.5 µg and a recommended intake of 6 µg daily. A typical American diet provides about 20 µg daily. Virtually all dietary cobalamin comes from meat or dairy products.
Absorption and metabolism of cobalamin. In humans, the uptake and transport of vitamin B12 (cobalamin) is a multi-step process, starting in the stomach, where the acidic environment facilitates the release of cobalamin from dietary proteins; this allows cobalamin to bind to a group of cobalamin-binding glycoproteins (salivary haptocorrin or "R binder”) that are produced in saliva.
Abbreviations: Ado-VitB12, adenosylcobalamin, also more fully 5′-deoxyadenosylcobalamin; Cubam R, cubam receptor, a multi-ligand receptor located in the terminal ileum, specializing in absorption of vitamin B12; FA, folic acid;...
The haptocorrin group of immunologically cross-reacting proteins includes transcobalamins I and III. The glycosylated structure of haptocorrin protein confers resistance to the acidic environment of the stomach (104).
The presence of food in the stomach also stimulates the secretion of "intrinsic factor," a glycoprotein produced by gastric parietal cells. In the neutral pH of the duodenum, intrinsic factor displaces salivary haptocorrin with the aid of pancreatic enzymes. The "cobalamin-intrinsic factor" complex protects cobalamin from destruction until it passes through the small intestine.
In the terminal ileum, the cobalamin-intrinsic factor complex is specifically recognized by the cubam receptor on the apical surface of enterocytes, triggering receptor-mediated endocytosis. The complex is transported to lysosomes for further processing, and the cubam receptor is recycled (79). Cubam is a multifaceted complex that is comprised of two essential components, cubilin (CUB) and amnionless (AMN) (196; 203): cubilin is a cell receptor, which is essential for recognizing the " cobalamin-intrinsic factor" complex, whereas amnionless is involved in the receptor mediated endocytosis of the complex into enterocytes in the ileum. Once the cobalamin-intrinsic factor complex reaches the lysosomes, intrinsic factor undergoes degradation, thereby releasing free cobalamin, which is then actively transported into the cytoplasm with the help of two transmembrane proteins (58). In the cytoplasm, free cobalamin is conveyed through the basolateral side of enterocytes via active transport (mediated by a multi-specific membrane transporter, multidrug-resistant protein 1, MRP1/ABCC1) or passive transport to the blood flow.
In the blood, cobalamin can bind with varying degrees of affinity to the known carriers, transcobalamin (previously named transcobalamin II) and haptocorrin. Cobalamin bound to transcobalamin (forming holotranscobalamin) represents the circulating bioavailable form of vitamin B12, which is subsequently absorbed by peripheral cells via endocytosis mediated by the receptor CD320 (also known as the transcobalamin II receptor), a ubiquitous cell surface receptor of the LDLR family (10). Following receptor-mediated endocytosis within lysosomes of peripheral target cells, transcobalamin is degraded, the CD320 receptor is recycled to the plasma membrane, and cobalamin, no longer complexed to a protein, can enter the cytoplasm via lysosomal transporters for utilization by the enzymes methionine synthase (MTR) and L-methylmalonyl-CoA mutase (MUT) (and may even be exported) (179).
In the blood, about 80% of cobalamin is bound to haptocorrin, forming holohaptocorrin, a complex that has a dual function: (1) holohaptocorrin conveys cobalamin to the liver, where it taken up through endocytosis (mediated by asialoglycoprotein receptor, ASGPR); and (2) cobalamin from holohaptocorrin can be transferred to transcobalamin, for which it has a higher affinity (163). Thus, holohaptocorrin is not only involved in the formation of the hepatic store of vitamin B12, but it is also itself a reserve of circulating vitamin B12 (163).
Metabolic consequences of cobalamin deficiency. In states of cobalamin deficiency, homocysteine and methylmalonic acid levels rise in the blood. How impairment of the two cobalamin-dependent reactions leads to the particular syndromes associated with deficiency is not completely understood. The hematologic and gastrointestinal abnormalities in cobalamin deficiency are similar to those of isolated folate deficiency and are generally attributable to impairment of DNA synthesis in the rapidly dividing cells of the gastrointestinal tract and bone marrow.
The failure of methionine synthesis due to cobalamin deficiency may either (1) lead to an accumulation of 5-methyl-tetrahydrofolate, trapping folate in a chemical form unusable in purine synthesis (the "folate trap" hypothesis) or (2) impair methylation reactions needed to produce formyl-tetrahydrofolate, a precursor to 5,10-methylene tetrahydrofolate. Whatever the mechanism, the impairment of DNA synthesis can be circumvented by the administration of sufficient amounts of exogenous folate. Thus, the hematologic and gastrointestinal effects of cobalamin deficiency are reversed by folate supplementation.
In contrast, it would appear that the neurologic effects of cobalamin deficiency are due to metabolic disturbances unrelated to purine synthesis because the neurologic deficits may develop independently of the hematologic abnormalities, and folate supplementation will not prevent or reverse these deficits. The exact mechanism of neurologic damage in vitamin B12 deficiency remains obscure, but disruption of normal myelin function appears to be important. Impaired methionine synthesis may lead to a depletion of S-adenosylmethionine, required for the synthesis of myelin phospholipids. Alternatively, accumulated methylmalonate and methylpropionate, precursors of the cobalamin-dependent synthesis of succinyl-CoA, may be incorporated abnormally into branched-chain fatty acids, resulting in abnormal myelination (88).
Causes of cobalamin deficiency. Disturbances at any step of cobalamin metabolism may result in actual or functional deficiency, but pernicious anemia accounts for the most cases of symptomatic vitamin B12 deficiency.
Pernicious anemia. Pernicious anemia is the result of an autoimmune gastritis in which the principal antigenic target is the H/K ATPase of gastric parietal cells. Gastric infection with H. pylori may trigger the underlying autoimmune gastritis (253). The destruction of parietal cells removes the source of both gastric acid and intrinsic factor, resulting in both gastric achlorhydria and pernicious anemia, respectively. The loss of intrinsic factor markedly reduces the efficiency of vitamin B12 absorption from food, which eventually leads to physiologically significant vitamin B12 deficiency. However, in the absence of any dietary cobalamin absorption, physiologic deficiency may not develop for 2 to 5 years because the liver stores about 3 mg of the vitamin.
Vitamin B12 deficiency as a manifestation of pernicious anemia may occur as part of an autoimmune polyglandular syndrome, frequently in association with type 1 diabetes mellitus, Hashimoto thyroiditis or hypothyroidism, and vitiligo (34; 243; 258; 223; 206; 274; 117).
Other gastrointestinal disorders. Besides pernicious anemia, various other iatrogenic conditions or naturally acquired gastrointestinal disorders can cause vitamin B12 deficiency.
Food-cobalamin malabsorption. A common cause of low vitamin B12 levels (but not necessarily of symptomatic deficiency) is the condition known as “food-cobalamin malabsorption,” in which vitamin B12 in food is absorbed poorly, but the crystalline vitamin B12 in vitamin pills is well absorbed (67; 48). Food-cobalamin malabsorption is associated with type B atrophic gastritis, a common condition that is associated with Helicobacter pylori infection. If gastric production of acid is reduced without concomitant loss of intrinsic factor (eg, as a result of H. pylori infection or medications), cobalamin absorption from food may be impaired (because the lack of the normal acidic environment of the stomach impedes the release of cobalamin from dietary proteins), but unbound cobalamin (eg, in supplemental vitamin preparations) will still be absorbed normally.
Bariatric surgery. Bariatric surgery may provide an anatomic and physiologic reason for vitamin B12 deficiency (160; 114; 82; 202; 157; 53). Both purely restrictive (eg, banding) and malabsorptive (eg, Roux-en-Y bypass) procedures increase the risk of vitamin B12 deficiency, although the malabsorptive procedures are more likely to do so (82). A deficiency in vitamin B12 is the most common nutritional deficiency in patients who have had gastric bypass surgery (114), but deficiencies of folate, pyridoxine, thiamine, vitamin D, copper, zinc, iron, and calcium may also occur (202; 157). Without supplementation, 30% of patients with Roux-en-Y gastric bypass develop vitamin B12 deficiency (160). Even with supplementation (with good adherence to prescribed supplements), a significant proportion of men (10%) and women (17%) develop vitamin B12 deficiency (157).
Gastrectomy. Gastrectomy for any reason (eg, gastric cancer, bariatric surgery) increases the risk of deficiency because parietal cells, the source of intrinsic factor, are removed (17; 26; 53; 255; 267; 233). In a meta-analysis of 14 studies of gastrectomy for gastric cancer, including collectively 2627 subjects, the prevalence of vitamin B12 deficiency surgery was 49% (26). Proximal gastrectomy is associated with a lower incidence of anemia and vitamin B12 deficiency compared to total gastrectomy in patients with upper gastric cancer (233).
Pancreatic insufficiency. Patients with pancreatic insufficiency may have reduced cobalamin absorption (because pancreatic enzymes normally assist with the displacement of salivary haptocorrin binding protein by intrinsic factor), although such patients rarely become cobalamin deficient.
Bacterial overgrowth syndrome. Bacterial overgrowth syndrome is a disorder in which poor intestinal motility allows certain intestinal bacteria to grow excessively, causing diarrhea and malabsorption. Bacterial overgrowth syndrome may cause cobalamin deficiency because of competition for cobalamin in the intestine before the "cobalamin-intrinsic factor" complex reaches the terminal ileum.
Fish tapeworm infection. Fish tapeworm infection may cause cobalamin deficiency because of competition for cobalamin in the intestine before the "cobalamin-intrinsic factor" complex reaches the terminal ileum. This process served as a plot point (albeit markedly dramatized) for an episode titled "Insensitive" of the TV show House that aired on February 13, 2007.
Diphyllobothrium latum, a species of fish tapeworm, is the largest tapeworm that can infect people, reaching lengths of up to 30 feet long; because the tapeworm absorbs approximately 80% of dietary vitamin B12, prolonged infection causes vitamin B12 deficiency and megaloblastic anemia in about 40% of cases.
Diphyllobothrium latum spends a portion of its life cycle in crustaceans and fish. An adult tapeworm in a human intestine releases unembryonated eggs in feces, where they can be identified on microscopic analysis. In water, this develops into an embryonated egg, from which a coracidium hatches. Coracidia are ingested by crustaceans, the first intermediate host, and then develop into procercoid larvae in the body cavities of the crustaceans. When infected crustaceans are ingested by fish, the procercoid larvae are released, and the last larval stage, a plerocercoid, develops in the muscle of the fish. When infected small fish are eaten by larger predator fish, the plerocercoid invades the tissue of the larger fish. The plerocercoid is the infective stage for man and other definitive hosts, including many fish-eating mammals and birds. When someone eats raw or inadequately cooked fish, usually from the Northern Hemisphere (although cases have also been reported in Uganda and Chile), the scolex of the plerocercoid attaches to the human intestine. The adult tapeworm develops from this. Diagnosis is made by identification of eggs or segments of the tapeworm (ie, proglottids) in a stool sample with a microscope. Adequately freezing or cooking fish will kill the parasite.
Pathology of the distal ileum. Any pathology of the distal ileum, where vitamin B12 is normally absorbed, may impair absorption of the intrinsic factor-cobalamin complex. Thus, cobalamin deficiency has been reported with regional enteritis, Whipple disease, ileal tuberculosis, tropical sprue, and surgical resection of the distal ileum, among other conditions. Although inflammation of the terminal ileum is common in Crohn disease and ulcerative colitis, some studies report that vitamin B12 deficiency is uncommon in these populations (30), whereas others report rates of vitamin B12 deficiency as high as 14% among patients with Crohn disease (30; 166) and 18% among patients with intestinal Behcet disease (an intestinal invasion of Behcet disease with chronic relapsing multisystem vasculitis disorder) (193).
Dietary cobalamin deficiency. Because no vegetable product is a reliable source of biologically active cobalamin, vegan diets can lead to cobalamin deficiency (132; 98; 83; 195; 39; 90; 181; 110). Compared with patients with pernicious anemia, strict vegetarians (who produce intrinsic factor normally) retain more cobalamin from the enterohepatic circulation and, therefore, may take much longer (as long as 10 to 20 years) to become symptomatic from cobalamin deficiency. Certain plant sources (eg, algae, mushrooms, fermented vegetables, and fermented beans) are potentially viable vitamin B12 sources for vegetarians, but this will require further study before a vegan diet can be considered to supply adequate vitamin B12 amounts (288). Despite this, cobalamin deficiency is common in vegans, occurring in up to half of such individuals who do not receive supplementation with vitamin B12 (132; 98; 83; 195). In many populations, vegans now receive appropriate supplementation (90; 110).
Because the breast milk of mothers who are on predominantly vegetarian diets may be deficient in vitamin B12, especially but not exclusively in resource-poor countries, exclusive breastfeeding can result in B12 deficiency in the infant (119; 152; 154; 39; 69; 75; 241; 228). This is a major causative factor for the high rate of vitamin B12 deficiency among infants and toddlers in India (121; 119). Infantile deficiency of vitamin B12 can affect neurodevelopmental outcome and cause spells, tremor, or irritability. Infantile B12 deficiency is characterized by developmental delay, sparse hair, hyperpigmentation, and tremors (135). Curiously, when treated with injectable B12, the affected babies can develop a peculiar transient "batwing dystonia" that may represent a "nutritional recovery" movement disorder (135). Acquired dietary vitamin deficiencies in infants may display reversible clinical symptoms mimicking inherited metabolic disorders (69). Concordant clinical presentation, suggestive neuroimaging findings, and/or biochemical evidence raise suspicion for the correct diagnosis, but acute neurologic conditions consistent with vitamin B12 deficiency should be treated without waiting for definitive biochemical confirmation (69). Preventing B12 depletion in newborns lowers healthcare costs and likely improves their health outcomes (75).
Lacto-vegetarians are also at increased risk of vitamin B12 deficiency (144). A lacto-vegetarian (sometimes referred to as a "lactarian") diet is a diet that requires abstinence from the consumption of meat and eggs while allowing consumption of dairy products.
Rarely, individuals with psychiatric conditions, including autism spectrum disorder, have prolonged histories of selective eating, sufficient to impair vitamin B12 absorption over periods of at least 7 to 10 years (219).
Autoimmune disorders (other than pernicious anemia). Transcobalamin receptor antibodies (anti-CD320) in autoimmune central vitamin B12 deficiency can produce impaired cellular uptake of cobalamin (B12) in vitro by depleting its target from the cell surface (200). In an affected woman with progressive tremor, ataxia, and scanning speech, B12 was nearly undetectable in her CSF despite a normal serum concentration. A patient-derived monoclonal antibody impaired B12 transport across an in vitro model of the blood-brain barrier. Immunosuppressive treatment and high-dose systemic B12 supplementation were associated with increased B12 in the CSF and clinical improvement.
Autoantibodies targeting the same epitope of CD320 were identified in seven other patients with neurologic deficits of unknown etiology, 6% of healthy controls, and 21% of a cohort of patients with neuropsychiatric lupus (200). In addition, in 132 paired serum and CSF samples, detection of anti-CD320 in the blood predicted B12 deficiency in the central nervous system even though these individuals did not display any hematologic signs of B12 deficiency (200). The authors found that the low-density lipoprotein receptor serves as an alternative B12 uptake pathway in hematopoietic cells but not in the nervous system (200).
Inborn errors of cobalamin metabolism. Several inherited conditions can cause actual or functional vitamin B12 deficiency. Two inherited disorders cause hereditary cobalamin deficiency: the Imerslund-Grasbeck syndrome and congenital pernicious anemia (intrinsic factor deficiency).
Imerslund-Grasbeck syndrome. The Imerslund-Grasbeck syndrome is a rare autosomal recessive condition characterized by both a specific congenital deficiency in cobalamin absorption and proteinuria (248). The syndrome can be caused by mutations in either of the two proteins, cubulin and amnion-associated transmembrane protein (amnionless), which together form the receptor complex called "cubam," needed for cobalamin absorption from the ileum. Cubilin is the intestinal receptor for the endocytosis of the "cobalamin-intrinsic factor" complex, whereas amnionless is a transmembrane protein. Cubilin is coded by the CUBN gene on chromosome 10p13, and amnionless is coded by the AMN gene on chromosome 10q32.32. The "cubam" complex also forms a protein reabsorption complex in the proximal renal tubule.
Congenital pernicious anemia (intrinsic factor deficiency). Gastric intrinsic factor is a fucosylated protein needed for B12 absorption (56). Intrinsic factor deficiency, or "congenital pernicious anemia," is a rare autosomal recessive disorder characterized by the lack of gastric intrinsic factor in the presence of normal gastric acid secretion and mucosal cytology (ie, without gastritis) and in the absence of intrinsic factor antibodies found in the acquired form of pernicious anemia. Hereditary intrinsic factor deficiency is caused by homozygous or compound heterozygous mutation of the CBLIF (cobalamin binding intrinsic factor) gene on chromosome 11q12.1 (168; 247; 248).
A heterozygous mutation in CBLIF (290T-> C) combined with the FUT2 (fucosyltransferase 2) secretor variant has been reported in children with congenital gastric intrinsic factor deficiency, impacting both gastric intrinsic factor secretion and B12 binding activity.
Neural tube defects. In addition to congenital gastric intrinsic factor deficiency, a heterozygous mutation in CBLIF (290T-> C) combined with the FUT2 (fucosyltransferase 2) secretor variant also produces severe forms of neural tube defects, suggesting that vitamin B12 delivery to neural tissue by the CUBN-gastric intrinsic factor pathway could play a role in the neural tube closure mechanisms (91).
Vitamin B12-responsive developmental and epileptic encephalopathy (mutation in the FUT2 gene). Understanding of the role of the FUT2 (fucosyltransferase 2) gene on chromosome 19q13.33 in determining vitamin B12 levels has developed since 2008 (93; 94; 245; 150; 56; 249; 259; 91; 28). Common variants of FUT2 are associated with plasma vitamin B12 levels (93; 94; 245; 150; 249; 282). The FUT2 secretor variant worsens B12 status in cases with heterozygous gastric intrinsic factor mutations by impairing gastric intrinsic factor secretion (56).
The FUT2 secretor variant p.Trp154Ter influences serum vitamin B12 concentration via holo-haptocorrin, but not holo-transcobalamin, and is associated with haptocorrin glycosylation (259). Transcobalamin is the bioactive form of the vitamin and is taken up by all tissues, whereas haptocorrin is only taken up by the liver.
Congenital transcobalamin II deficiency. Congenital transcobalamin II deficiency may cause megaloblastic anemia and neurologic abnormalities despite normal serum cobalamin levels (105). Transcobalamin II deficiency is an autosomal recessive disorder due to homozygous or compound heterozygous mutations in the TCN2 gene on chromosome 22q12.2. The clinical onset of transcobalamin II deficiency is in early infancy. The disorder is characterized by failure to thrive, megaloblastic anemia, pancytopenia, methylmalonic aciduria, recurrent infections, and vomiting and diarrhea. Treatment with high doses of parenteral methylcobalamin results in clinical improvement, but if left untreated, the disorder may result in intellectual disability and neurologic abnormalities (105).
Cobalamin defects or diseases A-H. Various inherited defects in the synthesis of the biologically active forms of vitamin B12 (ie, adenosylcobalamin and methylcobalamin) from ingested cobalamin substrate are referred to as cobalamin defects or diseases A-H; these usually present in infancy, although neurologic and psychiatric presentations of cobalamin C defect have been described in adolescents and adults (216; 43; 286; 06; 120). Cobalamin defects A, B, and H are characterized by isolated increased levels of methylmalonic acid in the blood and urine (216); most of these patients present in infancy with recurrent episodes of ketoacidosis without megaloblastic anemia. Cobalamin defects E and G are characterized by hyperhomocysteinemia and hypomethioninemia without methylmalonic aciduria (216); most patients with these diseases present in the first months of life with megaloblastic anemia, poor feeding, and, if not promptly diagnosed, various neurologic deficits, such as abnormal muscle tone and seizures. Cobalamin defects C, D, and F are due to defective synthesis of both methylcobalamin (resulting in hyperhomocysteinemia and hypomethioninemia) and adenosylcobalamin (resulting in methylmalonic aciduria) (216); most of these patients present in the neonatal period with feeding difficulties, failure to thrive, neurologic deterioration, megaloblastic anemia, variable renal and liver failure, cardiomyopathy, pneumonia, and retinopathy.
The metabolic pathways involved in remethylation disorders are complex (101).
In addition, the age of onset is quite wide, even if most cases have onset in infancy. The clinical presentations vary by age (see Table 1) (101).
|
Neonates (0 to 28 days) | ||
|
Encephalopathy | ||
|
Lethargy, apnea | ||
|
Nystagmus | ||
|
Infants (1 to 12 months) | ||
|
Growth failure or poor weight gain | ||
|
Muscular hypotonia | ||
|
Visual inattention or nystagmus | ||
|
Children (1 to 12 years) | ||
|
Chronic encephalopathy | ||
|
Muscular hypotonia or spasticity | ||
|
Acute progressive encephalopathy or apnea | ||
|
Paresthesia | ||
|
Hemolytic uremic syndrome | ||
|
Recurrent venous thrombosis | ||
|
Pulmonary hypertension | ||
|
Adolescents and adults (older than 12 years) | ||
|
Chronic encephalopathy | ||
|
Developmental disability, regression, or dementia | ||
|
Acute progressive encephalopathy | ||
|
Paresthesia | ||
|
Progressive limb weakness (legs> arms) | ||
|
Recurrent venous thrombosis | ||
|
Pulmonary hypertension | ||
|
| ||
Furthermore, multiple conditions mimic intracellular remethylation disorders, complicating the differential diagnosis of inborn errors of metabolism presenting with hyperhomocysteinemia (see Tables 2 and 3) (101).
|
Affecting cobalamin availability | |
|
Nutritional inadequacy (maternal vitamin B12 deficiency or vegan diet) | |
|
Affecting folate availability | |
|
Nutritional inadequacy (maternal deficiency or dietary inadequacy) | |
|
Other diseases with a combination of hematological and neurologic symptoms | |
|
Severe iron deficiency | |
|
| |
|
Macrocytosis or macrocytic anemia |
MMA |
Met |
Total vitamin B12 |
Folate | |
|
cblC | |||||
|
cblD-MMA/HC |
+ or − |
↗ |
↘↘ to nl |
nl |
nl |
|
cblF/cblJ |
+ |
↗ |
↘↘ to nl |
nl |
nl |
|
cblE/G |
+ |
nl |
↘↘ to nl |
nl |
nl |
|
cblD-HC |
+ or − |
nl |
↘↘ to nl |
nl |
nl |
|
MTHFR |
– |
nl |
↘↘ to nl |
nl |
nl or ↘ |
|
Vitamin B12 deficiency or malabsorption |
+ |
↗ |
↘ to nl |
↘↘ |
nl |
|
Folate deficiency or malabsorption |
+ |
nl |
↘ to nl |
nl |
↘↘ |
|
HCFC1 (cblX) |
+ or − |
↗ or nl |
↘↘ to nl |
nl |
nl |
|
CBS deficiency |
– |
nl |
nl-↗ |
nl |
nl |
|
TC deficiency |
+ |
↗ |
↘↘ to nl |
nl (↘) |
nl |
|
MTHFD1 deficiency* |
+ |
nl |
↘↘ to nl |
nl |
Nl |
|
From: (101) | |||||
Huemer and colleagues have developed a diagnostic and management pathway for patients with a suspected remethylation disorder and have provided a comparison of the various cobalamin defects associated with hyperhomocysteinemia, the respective enzymatic/incorporations tests available, and the genes involved in these diseases (see Table 4) (101).
Legend: Met, methionine; MMA, methylmalonic acid; MTHFD1, methylenetetrahydrofolate dehydrogenase 1; MTHFR, methylenetetrahydrofolate reductase; N, normal; OHCbl, hydroxotranscobalamin; TC, transcobalamin VitB12, tHcy, total ho...
|
cblC |
cblD-MMA/HC |
cblF |
cblJ |
cblD-HC |
cblE |
cblG |
MTHFR | |
|
Direct enzyme assay (tissues) |
no |
no |
no |
no |
no |
yes |
yes |
yes |
|
fib/leuc/amn |
fib/leuc/amn |
fib/leuc/amn | ||||||
|
Indirect enzyme assays | ||||||||
|
Propionate incorporation |
↘ |
↘ |
↘ |
↘ |
nl |
nl |
nl |
nl |
|
MTHF incorporation |
↘ |
↘ |
↘ |
↘ |
↘ |
↘ |
↘ |
nl |
|
Formate incorporation into serine |
↘ |
↘ |
↘ |
↘ |
↘ |
↘ |
↘ |
nl or ↗ |
|
Formate incorporation into methionine |
↘ |
↘ |
↘ |
↘ |
↘ |
↘ |
↘ |
↘ |
|
AdoCbl biosynthesis |
↘ |
↘ |
↘ |
↘ |
nl |
nl |
nl |
nl |
|
MeCbl biosynthesis |
↘ |
↘ |
↘ |
↘ |
↘ |
↘ |
↘ |
↘ |
|
Gene |
MMACHC |
MMADHC |
LMBRD1 |
ABCD4 |
MMADHC |
MTRR |
MTR |
MTHFR |
|
Chromosome location |
1p34.1 |
2q23.2 |
6q13 |
14q24.3 |
2q23.2 |
5p15.31 |
1q43 |
1p36.22 |
|
Mode of inheritance |
AR |
AR |
AR |
AR |
AR |
AR |
AR |
AR |
|
OMIM |
609831 |
611935 |
612625 |
603214 |
611935 |
602568 |
156570 |
607093 |
|
From: (101) | ||||||||
Despite treatment, many complications may occur from these disorders (see Table 5).
|
Growth and physical features |
Prenatal growth retardation and postnatal failure to thrive |
|
CNS |
Microcephaly |
|
Eye |
Nystagmus |
|
Blood |
(Macrocytic) anaemia |
|
Vascular |
Stroke |
|
Renal |
Haemolytic-uremic syndrome |
|
Heart |
Congenital heart defects |
|
| |
Huemer and colleagues have also provided guidelines for treating these disorders (see Table 6) (101).
|
Drugs with proven clinical effect |
Treatments without proven clinical effect |
To be avoided | |
|
Cobalamin-related remethylation disorders |
OHCbl parenteral |
Folate/folinic acid |
Nitrous oxide |
|
Betaine |
L-Carnitine |
Protein restriction | |
|
Methionine* | |||
|
MTHFR deficiency |
Betaine |
Folinic acid/5-Methylfolate* |
Nitrous oxide |
|
L-Carnitine |
Folic acid | ||
|
Methionine* |
Protein restriction | ||
|
From: (101) | |||
Treatment should be initiated with parenteral hydroxocobalamin without delay in any suspected remethylation disorder because it significantly improves survival and incidence of severe complications (101).
Cobalamin C defect or deficiency. Cobalamin C defect or deficiency is the most common inborn error of cobalamin metabolism, with an estimated prevalence of 1:200,000 births (06). Cobalamin C defect causes the accumulation of methylmalonic acid and homocysteine and decreased methionine synthesis. The gene responsible for the cobalamin C defect is the MMACHC, located on chromosome 1p34.1 (147). Cobalamin C defect causes impaired conversion of dietary vitamin B12 into its two metabolically active forms, methylcobalamin and adenosylcobalamin (164). The gene product catalyzes the reductive decyanation of cyanocobalamin. Adult cases are usually compound heterozygous carriers of a truncating and a nontruncating variant in the MMACHC gene (120).
Although the age of onset varies from prenatal to adult, most patients present in infancy. In infancy, cases have combined methylmalonic aciduria and homocystinuria. Early-onset cases of cobalamin C defect present with multisystem disease within the first year, with severe neurologic (eg, lethargy, hypotonia, microcephaly, seizures, stroke, neurodevelopmental delay), ocular (eg, rapidly progressing maculopathy with severe photoreceptor and ganglion cell loss), hematological, renal (eg, atypical hemolytic-uremic syndrome, renal arteriolar and glomerular thrombotic microangiopathy), gastrointestinal (eg, vomiting, diarrhea, feeding difficulties), cardiac, and pulmonary manifestations (eg, apneic episodes) (147; 42; 146; 43; 286; 118; 276; 03; 07; 29). Neonatal screening and early treatment can potentially improve the prognosis at least somewhat (286); but even with neonatal screening, outcomes are poor despite early initiation of therapy and regardless of the dietary strategy used (05).
In a study of 33 cases from Pakistan, the median ages of symptom onset, diagnosis, and treatment initiation were 300 (IQR:135-1800),1380 (IQR: 240-2730), and 1530 (IQR: 240-2790) days, respectively (03). The most common clinical features were cognitive impairment (88%), seizures (70%), motor developmental delay (61%), hypotonia (52%), and sparse/hypopigmented scalp hair (48%). The median pre-treatment HCY levels were 134 (IQR: 87.2-155.5) compared to post-treatment levels of 33.3 (IQR: 27.3-44.95) umol/L.
Despite the predominance of early-onset cases, more than 30 cases of adolescent-onset (12 to 17 years) and more than 30 cases of adult-onset (18 years or older) cobalamin C defect have been reported (06). Late-onset cases generally have a milder clinical phenotype with acute or slowly progressive neurologic symptoms and behavioral disturbances (06).
At the time of diagnosis, brain MRI showed diffuse white matter lesions in both hemispheres (T2) with diffusion restriction (A and B), and spinal MRI revealed subacute combined degeneration of the cord (C). After 42 months of t...
Adolescent cases may have autism spectrum disorder, mental retardation, rapidly progressing maculopathy with severe photoreceptor and ganglion cell loss, and movement disorders (42; 286; 178). Rapidly progressing maculopathy with severe photoreceptor and ganglion cell loss, renal thrombotic microangiopathy, hemolytic uremic syndrome, pulmonary hypertension, and pulmonary thrombotic events have also been described in adult-onset cobalamin C defect (42; 06; 120).
Adult patients with cobalamin C defect pose a diagnostic challenge because they typically present with normal to high-normal serum vitamin B12 concentrations without macrocytic anemia (06; 120). Cobalamin C defect should be considered in the differential diagnosis in adult patients with atypical hemolytic uremic syndromes, slow unexplained decline in renal function, idiopathic neuropathies, spinal cord degenerations, ataxias, unexplained or recurrent thrombosis, visual field defects, maculopathy, and optic disc atrophy (120). Plasma homocysteine measurement should be obtained when the disease is suspected (101; 120). To further aid diagnosis, genes belonging to the intracellular cobalamin pathway should be routinely included in gene panels for atypical hemolytic uremic syndrome and chronic kidney disorders.
Treatment should be initiated with parenteral hydroxocobalamin without delay in cases of cobalamin C defect, as in any suspected remethylation disorder, because it significantly improves survival and incidence of severe complications (101). In adult cases, treatment with intramuscular hydroxycobalamin was effective in reversing symptoms (120; 184). Earlier recommendations advocated treatment of the cobalamin C defect with a combined approach that utilizes vitamin B12 plus betaine to provide a substrate for the conversion of homocysteine into methionine through betaine-homocysteine methyltransferase, and folic acid to enhance the remethylation pathway (164). It is unclear if these additional measures change clinical outcomes. No proven efficacy has been demonstrated for carnitine and dietary protein restriction (164).
Despite these measures, the long-term outcomes are unsatisfactory, especially in cases with early onset, with frequent progression of neurologic and ocular impairment (164; 43).
Medication-induced cobalamin deficiency. Medications are not an uncommon etiology of vitamin B12 deficiency.
Nitrous oxide. Nitrous oxide irreversibly oxidizes the cobalt in cobalamin, which effectively inactivates vitamin B12.
Thus, a functional deficiency of cobalamin with associated neurologic manifestations can be created by the inhalation of nitrous oxide, a common anesthetic agent and recreational drug (60; 61; 112; 122; 133; 145; 201; 02). Nitrous oxide abusers can have non-B12-related psychiatric problems, as might be expected for substance abusers in general (194). Nitrous oxide abuse may cause more severe clinical symptoms, peripheral nerve damage, and spinal cord injury than is commonly seen with other forms of vitamin B12 deficiency (02).
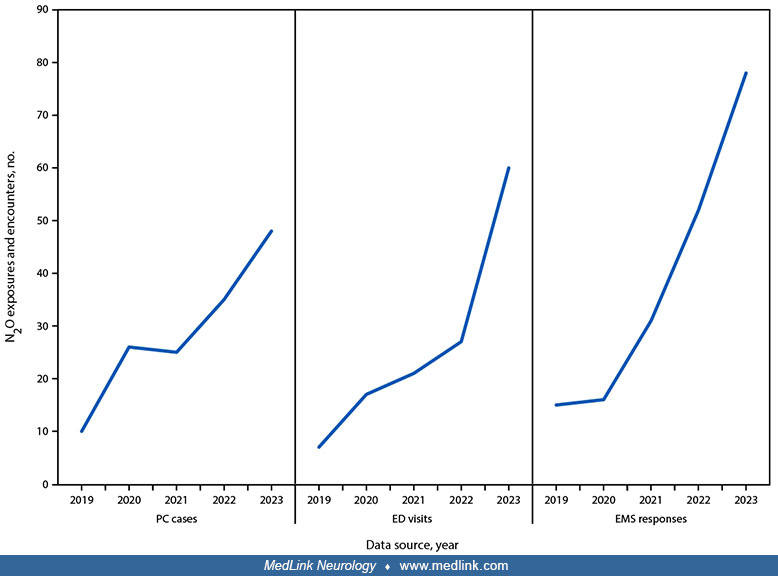
The frequency of nitrous oxide abuse has been increasing. In Michigan in 2023, annual poison center exposures, emergency department visits, and emergency medical service responses related to nitrous oxide misuse were four to five times more than those seen in 2019 (264).
Poison center cases, emergency department visits, and emergency medical service responses related to recreational nitrous oxide misuse -- Michigan, 2019 to 2023. Abbreviations: ED = emergency department; EMS = emergency medical...
Proton pump inhibitors or H2-receptor blocking agents. Proton pump inhibitors and H2-receptor-blocking agents have been implicated as causes or contributors to vitamin B12 deficiency because they can produce iatrogenic gastric achlorhydria (116; 106). A decrease in serum cobalamin levels has been demonstrated after 3 to 4 years of omeprazole therapy.
Metformin. Diabetics on metformin are also at risk for vitamin B12 deficiency because the drug impairs vitamin B12 absorption (12; 35; 100; 126; 13; 142; 284; 115; 222). The mechanism of this effect has been debated (252; 14), and the relationship between metformin exposure and vitamin B12 deficiency remains somewhat ill-defined (192; 214; 281). Several randomized, placebo-controlled trials indicate that metformin therapy leads to a progressive decline in vitamin B12 levels and can cause physiologically significant deficiency (153). Metformin-related vitamin B12 deficiency may cause or accelerate distal symmetrical and autonomic neuropathy in the patient with diabetes (35). Patients randomized to metformin therapy in the Diabetes Prevention Program Outcome Study were more likely to show vitamin B12 deficiency or borderline low vitamin B12 levels (19). Populations already vulnerable to vitamin B12 deficiency, such as the institutionalized elderly, may be at especially heightened risk of deficiency in the setting of metformin use (275). Although routine B12 supplementation in patients on metformin therapy is not the standard of care (279), it is reasonable to monitor vitamin B12 levels in patients on metformin, particularly for individuals on long-term metformin (102; 103) or high-dose metformin (≥1500 mg/day) (81; 103), both of which are risk factors for metformin-associated vitamin B12 deficiency (81; 102; 103). Certainly, the development of peripheral polyneuropathy in a diabetic patient on metformin should not be assumed to be due to diabetes alone without checking a vitamin B12 level.
A previously unappreciated mechanism may explain the relationship between metformin use and vitamin B12 deficiency. Apparently, metformin acts directly on gut bacteria to increase their vitamin B12 uptake, leading to increasing competition for available dietary vitamin B12 and ultimately to human vitamin B12 deficiency (55). Bacteroides ovatus abundance was negatively correlated with serum levels of vitamin B12 in subjects with type 2 diabetes who received metformin treatment. Bacteroides ovatus accumulates cobalamin due to BtuB upregulation; BtuB, a bacterial outer membrane vitamin B12 receptor/transporter, transports vitamin B12 across the outer membrane of gram-negative bacteria. Upregulation of this transporter effectively sequesters dietary vitamin B12 so it cannot be absorbed from the gut. The metabolic effects of metformin vary somewhat across gram-negative bacteria: (1) in E coli metformin inhibits respiratory chain complex I, impairing the function of the inner membrane vitamin B12 transporter (BtuCD), so vitamin B12 entry is blocked at the inner membrane, causing vitamin B12 to accumulate in the periplasm; (2) in contrast, in B ovatus metformin upregulates respiratory chain complex V so that vitamin B12 can be transported across inner membrane to the bacterial cytoplasm (55).
In an experimental test of the effectiveness of this mechanism, B ovatus accelerated metformin-induced vitamin B12 deficiency in mice fed a high-fat diet.
Levodopa. Conflicting evidence links chronic levodopa therapy in patients with Parkinson disease to an increased risk of symptomatic vitamin B12 deficiency, perhaps because carbidopa metabolism through the catechol-O-methyl transferase pathway may deplete vitamin B12-dependent cofactors (204; 51; 172). Patients receiving continuous enteral levodopa infusions may be particularly at risk (172; 167). Checking vitamin B12 levels may be worthwhile before instituting levodopa therapy and also periodically in patients under chronic treatment.
|
• The elderly are at increased risk of cobalamin deficiency, with rates of laboratory-diagnosed deficiency near 15% in some studies. | |
|
• Both vegans and vegetarians have increased rates of vitamin B12 deficiency. | |
|
• Other groups at high risk include those with (1) HIV infection and AIDS; (2) malnutrition; (3) intestinal parasitism; and (4) Crohn disease, particularly those who have had an ileal resection. |
The elderly are at increased risk of cobalamin deficiency, with rates of laboratory-diagnosed deficiency near 15% in some studies.
Both vegans (those whose diets allow no animal products) and vegetarians (diets that can include some dairy or eggs but no meat) have high rates of functional vitamin B12 deficiency (98; 83; 195; 213). Of particular concern are the breastfed infants of vegetarian or vegan mothers and children on vegetarian or vegan diets (50).
Other groups at high risk include those with HIV infection and AIDS, malabsorption due to gastrointestinal illness (eg, Crohn disease, tapeworm infestation), as well as certain populations outside the United States where malnutrition and intestinal parasitism may play a role (16; 21). Persons with Crohn disease, particularly those who have had an ileal resection, are at increased risk of vitamin B12 deficiency (38).
In an Indian cross-sectional study of 100 children less than 5 years old with a diagnosis of severe acute malnutrition (according to WHO criteria), 45% were vitamin B12 deficient (less than 203 pg/mL) (21). Hyperhomocysteinemia (greater than14 μmol/L) was present in 39%, and among these, 69% had concomitant low serum vitamin B12 levels. Severe anemia and hypoproteinemia were significantly and independently associated with vitamin B12 deficiency. Of the 45 children who were vitamin B12 deficient, most had delays in gross motor (93%), fine-motor (87%), language (62%), and adaptive-cognitive (80%) abilities. Vitamin B12 level was significantly associated with developmental delay.
Conclusion. There is a high prevalence of vitamin B12 deficiency in children with severe acute malnutrition, which is also associated with development delay across all domains (except language) in these children.
Vitamin B12 deficiency and dementia. Many epidemiological investigations have reported an association between lower levels of vitamin B12 or folate (or elevated blood homocysteine and methylmalonic acid, which are metabolic indicators of vitamin B12 deficiency) and Alzheimer disease or other forms of cognitive impairment (57; 270; 239; 123; 131; 137). A community-based study of elderly persons similarly showed that serum indices of relative vitamin B12 levels were associated with increased risk of progressive brain atrophy over 5 years (262). Another study of elderly community-dwelling individuals found that low vitamin B12 levels were associated with poorer memory (175). In contrast, a Danish cohort study showed no association between low plasma vitamin B12 levels and dementia (18).
Studies in rodent models of Alzheimer disease demonstrate that both B-vitamin deficiency and hyperhomocysteinemia can increase levels of the amyloid-forming peptides A-beta 1-40 and A-beta 1-42, increase levels of beta-amyloid plaque, and cause cognitive impairment (190; 287; 289).
Despite the suggestive epidemiological and experimental data, no prospective trial has demonstrated that supplementation of the diet with vitamin B12 or folate can prevent or delay the onset of Alzheimer disease or other dementia. The cognitive performance of patients with mild or moderate B12 deficiency and dementia usually does not improve with vitamin B12 supplementation (49; 59; 138; 240; 73; 27). The VITALS trial randomly assigned 409 patients with clinically diagnosed Alzheimer disease (MMSE scores between 14 and 26) and normal vitamin B12, folate, and total homocysteine levels to daily B vitamin supplementation (B12 1 mg, B6 25 mg, folate 5 mg) or placebo, and followed the patients for 18 months (08). No effect on the rate of decline of cognitive function was observed.
Modest and inconclusive beneficial effects of vitamin B12 supplementation were observed in two trials. In an Australian study of the effect of vitamin B12, folate, and other randomized interventions on cognitive function in a group of subjects at increased risk for depression, an improvement in some memory tests was observed in those randomized to vitamin B12 and folate supplementation (268). A British group has found that vitamin B12 and folate supplementation reduced the rate of brain atrophy in persons with mild cognitive impairment, and this effect was particularly pronounced in those subjects with higher levels of homocysteine at baseline (231; 68). The effect on tests of cognition was not reported in detail.
The discrepancy between, on the one hand, repeated findings of an association between indices of relative vitamin B12 deficiency and cognitive impairment and, on the other hand, the lack of substantial benefit in therapeutic trials of B vitamin supplementation is not understood. Most clinical trials have been of relatively short duration, so it is possible that more substantial benefits would be seen with more prolonged therapy (95).
B vitamins and cerebrovascular disease. Observational studies have found a direct relation between mild or moderately elevated serum homocysteine levels and the risk of both coronary artery disease and cerebrovascular thrombosis (260; 285; 209). Mild to moderate elevations in homocysteine may also be associated with increased risk for cranial artery dissections (80). Cobalamin deficiency, which elevates plasma homocysteine, might, thus, be a cause of otherwise unexplained ischemic stroke or cranial artery dissection (197; 272). Total homocysteine levels are inversely related to the levels of both folate and cobalamin consumed in the diet (229), but in countries where grain products are fortified with folate, vitamin B12 intake may be the major determinant of homocysteine levels (89). Thus, cobalamin deficiency might increase the risk of cerebral and myocardial infarction by increasing serum homocysteine levels.
In a retrospective cross-sectional study of 165 patients with thromboembolic and other cardiovascular manifestations identified among 1006 hospitalized patients, 165 patients (84%) had at least moderate hyperhomocysteinemia with levels greater than 30 μmol/L, 27% had levels greater than 50 μmol/L, and 3% had severe elevations greater than 100 μmol/L (148). Hyperhomocysteinemia was related to vitamin B12 or folate deficiency in 55%, mutations in one or more genes of one-carbon or vitamin B12 metabolism in 11%, and severe renal failure in 15%. Hyperhomocysteinemia was the sole vascular risk identified in almost 9% of patients. Sixty percent received a vitamin supplement to treat hyperhomocysteinemia, with a significant decrease in median homocysteine levels. No recurrence of thromboembolic manifestations was observed after vitamin supplementation and antithrombotic treatment of patients who had hyperhomocysteinemia as an isolated risk factor after approximately 4 years of follow-up.
Conclusion. The high frequency of intermediate or severe hyperhomocysteinemia differs from the frequent moderate hyperhomocysteinemia reported in previous observational studies of patients with pre-existing cerebrovascular disease. Our study points out the importance of diagnosing and treating nutritional deficiencies and inherited disorders to reverse intermediate or severe hyperhomocysteinemia associated with cerebrovascular disease outcomes.
Despite these observational studies, the prospective, blinded, and placebo-controlled VISP, NORVIT, HOPE-2, WAFACS, VITATOPS, and SEARCH trials have all failed to show a significant benefit of high-dose B vitamin (including cobalamin) supplementation for preventing thrombotic vascular events or death (254; 41; 158; 11; 242; 261). These six large studies were similar in design. All examined the effect of combined folate and vitamin B12 supplementation (most with vitamin B6 or pyridoxine as well) on the rate of thrombotic vascular events in high-risk subjects. Subjects were recruited from patients with nondisabling cerebral infarction (VISP); myocardial infarction (NORVIT); cerebrovascular disease, peripheral vascular disease, or coronary disease or diabetes (HOPE-2); or female health professionals with known cardiovascular disease or vascular disease risk factors (WAFACS), recent stroke or transient ischemic attack (VITATOPS), or myocardial infarctions (SEARCH). Subjects were followed for 24 (VISP), 40 (NORVIT), 60 (HOPE-2), 88 (WAFACS), 41 (VITATOPS), or 80 months (SEARCH). No study demonstrated a significant reduction in the rate of coronary events or stroke. The dose of vitamin B12 used in the VISP and NORVIT trials and issues with the design of the VISP trial might have obscured a small but still meaningful benefit to vitamin B12 supplementation (234). A meta-analysis of patients from two large trials revealed that patients with normal kidney function may benefit from vitamin B12 supplementation for stroke prevention, whereas patients with impaired kidney function may not (235).
|
• In at-risk individuals, vitamin B12 deficiency can be prevented through oral supplementation with the vitamin. | |
|
• Vitamin B12 deficiency in pregnant and breastfeeding women needs to be recognized and corrected because of the potential for irreversible neurologic injury to the child. |
In those at risk (see Epidemiology section for a description of risk groups), vitamin B12 deficiency can be prevented through oral supplementation with the vitamin (see Management section).
In gastric cancer after gastrectomy (which removes the gastric parietal cells and, thus, the source of intrinsic factor needed for normal vitamin B12 absorption), supplementation with vitamin B12 can prevent the development of vitamin deficiency (17). In a trial involving 74 patients comparing vitamin B12 supplementation with 500 μg/day versus 1500 μg/day after 3 months of treatment, 8% of those on the lower dose had deficient levels versus none on the higher dose (17). Because no statistical significance was identified in this small trial, the authors incorrectly concluded that oral vitamin B12 500 μg/day supplementation is as effective and safe as oral vitamin B12 1500 μg/day supplementation in these patients. Based on the results obtained, it would be prudent to use doses of at least 1000 μg/day (standard dosing in the United States).
It is particularly important to be alert to possible vitamin B12 deficiency in pregnant and breastfeeding women because of the potential for irreversible neurologic injury to the child. Newborn screening for neonatal vitamin B12 deficiency may lead to a fourfold risk reduction of developing symptomatic vitamin B12 deficiency in the first year of life compared with infants without newborn screening (173).
The neurologic manifestations of vitamin B12 deficiency are not specific, and other causes of subacute to chronic neuropathy, myelopathy, or both will need consideration depending on the clinical scenario. A differential for the myelopathy would include various metabolic, toxic, immunologic, and infectious disorders, such as copper deficiency myelopathy (possibly precipitated by zinc toxicity), folate deficiency myelopathy, vitamin E deficiency myelopathy, nitrous oxide toxicity, HIV-related vacuolar myelopathy, HTLV-1-associated myelopathy, neurosyphilis, and possibly Lyme disease. Conditions with acute or episodic or relapsing-remitting courses are less likely to be confused with vitamin B12 deficiency myelopathy (eg, trauma, multiple sclerosis), as are myelopathies resulting from intramedullary lesions or extramedullary compression. In cases where peripheral neuropathy is the presenting feature, evaluation for common causes (eg, impaired glucose metabolism, hypothyroidism) is necessary, keeping in mind that autoimmune disorders can present with combinations of disorders (eg, pernicious anemia, autoimmune thyroid dysfunction, and type 1 diabetes).
The hematologic and gastrointestinal signs of vitamin B12 deficiency can be mimicked by folate deficiency (66; 207). Although the evidence for excess folic acid exacerbating vitamin B12 deficiency is weak, it is prudent to increase vigilance for identifying B12 deficiency in at-risk individuals, including older adults and others with low B12 intake or conditions that are associated with B12 malabsorption, who also ingest excessive folic acid or are prescribed folic acid in high doses (169).
A deficiency of vitamin B12 transport in the blood may rarely cause subacute combined degeneration in the presence of normal serum vitamin B12 levels (211).
Although rare, copper deficiency may present with subacute combined degeneration (134; 108). Copper deficiency may follow bariatric surgery or zinc ingestion. Until recently, zinc was present in high quantities in dental adhesives (109). Toxic zinc exposures may occur with ingestion of coins (eg, in schizophrenics and children) and in certain occupational exposures (140).
|
• Although there is no well-accepted “gold standard” for confirming vitamin B12 deficiency, there are two somewhat complementary methods of assessing vitamin B12 status: (1) directly measuring vitamin B12 levels in the serum and (2) measuring levels of metabolites (fasting serum homocysteine and serum methylmalonic acid levels) that accumulate as a result of inadequate vitamin B12. | |
|
• Many patients with neurologic signs of vitamin B12 deficiency do not have readily detectable hematological abnormalities. | |
|
• Traditionally, evaluation of possible vitamin B12 deficiency begins with measurement of the serum cobalamin level, although neurologically significant deficiency may occur with low-"normal" serum levels. | |
|
• In cases with borderline low serum cobalamin levels, measuring serum methylmalonic acid and fasting homocysteine levels may clarify whether these are truly low-normal values or whether these instead indicate a physiologically significant functional vitamin B12 deficiency. | |
|
• From a diagnostic standpoint, it is not sufficient to establish the presence of vitamin B12 deficiency; it is also necessary to determine why the cobalamin deficiency developed in the first place. | |
|
• A laboratory-supported clinical diagnosis of cobalamin deficiency, with hematologic or neurologic changes, implies the presence of long-standing gastrointestinal disease until proven otherwise. | |
|
• Simply treating the cobalamin deficiency does not address the underlying problem that brought about the cobalamin deficiency. | |
|
• Cobalamin deficiency should not be assumed to be due to pernicious anemia (ie, autoimmune destruction of parietal cells resulting in intrinsic factor deficiency) simply because that is the most common cause. | |
|
• A diagnosis of pernicious anemia can be made by demonstration in the serum of either anti-intrinsic factor antibodies or antibodies to gastric parietal cells in the setting of symptomatic vitamin B12 deficiency. However, the absence of such antibodies does not exclude the diagnosis of pernicious anemia. | |
|
• In the absence of confirmatory antibody studies establishing a diagnosis of pernicious anemia, gastroenterology consultation and other diagnostic tests (eg, endoscopy) may be necessary to determine the cause of cobalamin deficiency if a clear and sufficient explanation is not readily evident from the patient's history. |
Although there is no well-accepted “gold standard” for confirming vitamin B12 deficiency (92), there are two somewhat complementary primary methods of assessing vitamin B12 status: (1) directly measuring vitamin B12 levels in the serum and (2) measuring levels of metabolites that accumulate as a result of inadequate vitamin B12. In addition, supportive testing addresses hematological abnormalities, but hematological abnormalities should not be considered an adequate screening test for vitamin B12 deficiency (vide infra). Ancillary testing may include neuroimaging and electrophysiological tests. In some cases, the sequential use of different B12 status biomarkers or the calculation of a composite B12 status score (derived from a panel of B12 biomarkers and adjusted for folate status and age) can be used to detect deficient states that may otherwise be overlooked when using a single biomarker approach such as a B12 level (92).
Once vitamin B12 deficiency is established, it is critical to identify the underlying cause of the deficiency. Appropriate evaluation for this purpose includes antibody tests for pernicious anemia, consideration of gastroenterology consultation, and possibly endoscopy.
Hematological abnormalities. Many patients with neurologic signs of vitamin B12 deficiency do not have readily detectable hematological abnormalities, such as megaloblastic anemia (also known as macrocytic anemia) or neutrophils with hypersegmented nuclei (151; 96; 205; 266; 76). Therefore, clinicians need to recognize the neurologic symptoms and signs of vitamin B12 deficiency and specifically seek evidence for this diagnosis if it might explain a patient’s condition rather than waiting for hematologic evidence of vitamin B12 deficiency to turn up on a complete blood count ordered for that or other purposes.
The diagnostic utility of mean corpuscular volume (MCV), as an indicator of abnormal serum methylmalonic acid (MMA) and total plasma homocysteine (tHCY) biomarkers of vitamin B12 or folate deficiency, was assessed in 26,397 participants (12,730 males and 13,667 females) in the National Health and Nutrition Examination Survey (NHANES) dataset (76). A positive and significant correlation between MCV and these indicators was found in the general population between the ages of 18 and 85. In pregnant women, for every unit increase in MCV, there was a 19% increase in odds of abnormal methylmalonic acid, OR 1.19 (95% C.I. 1.08-1.31; p=0.001), and the Area Under the Curve for MCV as a test for abnormal methylmalonic acid was 78%. An MCV cutoff of 93.1 correctly identified abnormal methylmalonic acid in pregnant women with 81% sensitivity and 77% specificity. In the general, population MCV performed poorly in identifying abnormal MMA and tHCY. Mean corpuscular volume is an inexpensive measurement that may be useful to screen asymptomatic pregnant women for vitamin B12 abnormalities, which in turn may have a significant impact on reducing adverse neurologic outcomes in their children.
Although hypersegmented neutrophils (ie, neutrophils with five or more nuclear lobes) are most often associated with megaloblastic anemia (eg, folic acid or vitamin B12 deficiency, including pernicious anemia), they may also be seen in myelodysplastic syndromes, in patients receiving chemotherapy, or in long-term chronic infections. Even when present, hypersegmented neutrophils are easily missed on routine examination of the blood smear. Folate supplementation can normalize the hematologic abnormalities in vitamin B12 deficiency (thereby potentially obscuring or delaying diagnosis), but even patients who are not taking folate supplements may have no evident hematologic abnormalities despite symptomatic vitamin B12 deficiency.
Bone marrow aspirates may support the diagnosis of megaloblastic anemia, demonstrating erythroid hyperplasia and megaloblasts with sieve-like nuclear chromatin and megaloblasts with giant, abnormal-shaped stab forms (191).
Bone marrow aspirate smears demonstrating erythroid hyperplasia and megaloblasts with sieve-like nuclear chromatin (thick arrow). This is consistent with a diagnosis of megaloblastic anemia (May-Grünwald-Giemsa staining x400). ...
Bone marrow aspirate smears demonstrating erythroid hyperplasia and megaloblasts with giant, abnormal-shaped stab forms (thin arrow). This is consistent with a diagnosis of megaloblastic anemia (May-Grünwald-Giemsa staining x40...
Cobalamin levels. Traditionally, evaluation of possible vitamin B12 deficiency begins with measurement of the serum cobalamin level. Usual lower limits of normal are approximately 170 ng/L (111 pM/L) for the radioimmunoassay and 250 ng/L (184 pM/L) for the chemiluminescent assay. However, neurologically significant deficiency may occur with low-"normal" serum levels. In addition, serum vitamin B12 concentrations may vary with the concentrations of the binding proteins. Falsely increased values may be caused by myeloproliferative disorders or liver disease, whereas falsely low values can be seen with folate deficiency, pregnancy, multiple myeloma, and transcobalamin deficiencies. In some patients with myeloproliferative disorders or liver disease, increased levels of serum haptocorrins result in increases in the measured serum cobalamin level (without actually increasing the physiologically active holotranscobalamin fraction).
Diagnosis is further complicated by the fact that certain automated assays may indicate false normal or even elevated vitamin B12 levels in patients with high levels of anti-intrinsic factor antibodies, and such antibodies are found in 70% of patients with pernicious anemia (47; 280; 237).
Serum cobalamin levels do not strictly correlate with physiologic deficiency, in part because traditional assays measure total serum cobalamin, even though the fraction bound to transcobalamin II (holotranscobalamin II) is physiologically most important. Certain conditions, such as liver disease, can increase serum haptocorrin, thereby increasing total serum cobalamin, even with relatively low levels of cobalamin bound to transcobalamin II (232).
Holotranscobalamin II. Transcobalamin II transports cobalamin in plasma to receptors on cell membranes; only the fraction of cobalamin that is bound to transcobalamin II is biologically active. Although some investigators have advocated the measurement of serum holo-transcobalamin II as a theoretically better measure of active vitamin B12, its clinical superiority has not been established, and holo-transcobalamin II assays are technically difficult to perform (129; 182; 85; 257; 47).
Metabolites: methylmalonic acid and fasting homocysteine. In cases with borderline low serum cobalamin levels, measuring serum methylmalonic acid (MMA) and fasting homocysteine levels may clarify whether these are truly low-normal values or whether they instead indicate a physiologically significant functional vitamin B12 deficiency. In addition, if a patient’s clinical situation strongly suggests vitamin B12 deficiency (eg, based on characteristic neurologic abnormalities), MMA or fasting homocysteine levels should be determined, even if the lab reports a normal or even high vitamin B12 level. Testing of MMA has additional value in identifying patients with clinically relevant metabolic deficiency when vitamin B12 is below 304 pmol/L (412 pg/mL) (271).
MMA is somewhat more sensitive than fasting homocysteine as an indicator of cobalamin deficiency, but some vitamin B12-deficient patients have an isolated elevation of homocysteine (85). Defining the levels of MMA or homocysteine that are definitely pathologic is difficult. Traditionally, greater than three standard deviations above the mean is considered elevated, but levels less elevated than this will often decline in patients supplemented with cobalamin or folate, perhaps indicating a relative deficiency of these vitamins. The greater-than-3-standard-deviation cutoff for serum MMA is generally around 350 nM/L and for homocysteine is 15 µM/L (20 µM/L in patients over age 60 years). A practice guideline of the American Academy of Neurology recommends measuring MMA or fasting homocysteine levels in patients with unexplained distal symmetric polyneuropathy and low-"normal" vitamin B12 levels (71). A retrospective case series found that 10 of 23 patients with neuropathy, low-"normal' vitamin B12 levels, and elevated MMA levels improved with vitamin B12 supplementation (176).
Although methylmalonic acid is an important functional marker of vitamin B12 deficiency, vitamin B12 status is not the only determinant of MMA levels. Methylmalonic acid levels also relate to renal function, endogenous production of propionic acid, and the function of various genes (212). In fact, less than a quarter of the variation in MMA levels is explained by vitamin B12 levels, eGFR, age, and sex, indicating that a large part of the variation in MMA levels is attributable to other factors (eg, catabolism, dietary components, or gut microbial production). Correcting plasma MMA for eGFR can reclassify plasma MMA levels across decision limits for diagnosing B12 deficiency, particularly for patients with reduced kidney function (eg, grade 3 or worse chronic kidney disease) (180).
Higher MMA levels are associated with an increased risk for mortality, independent of vitamin B12 status and sex, but are more pronounced in individuals with impaired renal function (212).
Deficiency of vitamin B12 or folic acid can lead to increased homocysteine levels.
Ancillary studies. Several ancillary studies may help support the diagnosis and also serve as indicators of the severity of neurologic involvement.
Neuroimaging. MRI scans in patients with vitamin B12 deficiency may show nonspecific regional hyperintensities in the affected tracts or structures on T2-weighted images, but the sensitivity of MRI for detecting signs of vitamin B12 deficiency is low (107). Typically, these MRI abnormalities are found in the posterior columns, but abnormalities may also be found in the cerebral white matter and, rarely, in other locations, such as the globus pallidus (52; 141; 226). In a population-based cohort study, the burden of MRI T2 hyperintensities in the white matter correlated with biochemical indices of relative vitamin B12 deficiency, even when these indices were in the generally accepted normal range (63). In infants with vitamin B12 deficiency, MRI may show delayed myelination (86).
Electrophysiological studies. In electrophysiology studies, the characteristic abnormality of vitamin B12 deficiency in the peripheral nervous system motor is a length-dependent sensory axonopathy (77). Evidence of denervation is typically greater in sensory axons than in motor axons and is more severe distally than proximally. Conduction velocities are normal. Sensory evoked response studies may show prolonged L3-P27 intervals (indicating slowed conduction between the cauda equina and the somatosensory cortex of the brain). Visual-evoked responses may be normal or may show abnormalities in patients with subacute combined degeneration, even in the absence of visual symptoms (97; 77).
Megaloblastic changes in the bone marrow are usually seen in vitamin B12 deficiency, even in the absence of abnormalities in the indices of circulating erythrocytes.
Ascertaining the cause of cobalamin deficiency. From a diagnostic standpoint, it is not sufficient to establish the presence of vitamin B12 deficiency; it is also necessary to determine why the cobalamin deficiency developed in the first place. A laboratory-supported clinical diagnosis of cobalamin deficiency, with hematologic or neurologic changes, implies the presence of long-standing gastrointestinal disease until proven otherwise. Simply treating the cobalamin deficiency does not address the underlying problem that brought about the cobalamin deficiency.
Cobalamin deficiency should not be assumed to be due to pernicious anemia (ie, autoimmune destruction of parietal cells resulting in intrinsic factor deficiency) simply because that is the most common cause. Vitamin B12 deficiency may be the first sign of otherwise overlooked abnormalities of the gastrointestinal system. At an elementary level, it is important to obtain a careful dietary history, review medications thoroughly to identify any that may interfere with cobalamin absorption or metabolism, and obtain a thorough review of systems and past medical history to identify potential causes of cobalamin deficiency.
Cobalamin absorption testing (Schilling test). The Schilling test, which was developed in 1953, relied on radiolabeled cobalamin to track defects in absorption. This test was previously used to investigate and clarify the cause of cobalamin deficiency, but unfortunately, this test is not available in the United States (45). There were two parts to the test: (1) cobalamin malabsorption was identified by poor urinary excretion of an oral dose of cobalamin; (2) gastric pathologies were identified by demonstrating a significant increment in urinary excretion of an oral dose of cobalamin when administered in combination with an external source of intrinsic factor, whereas the absence of a significant increment under these circumstances indicated an intestinal pathology.
Unfortunately, the test proved to be too complicated and unwieldly for many patients and their clinicians as it required "an overnight fast, ingestion of (modestly) radioactive cobalamin, a 24-hour collection of urine, a cobalamin injection, two trips to the laboratory, and, if the result [was] abnormal, repetition of the entire procedure with a dose of [intrinsic factor]" (45). In addition, it required normal renal function and complete urine collection.
Antibody tests for pernicious anemia. A diagnosis of pernicious anemia can be made by demonstration in the serum of either anti-intrinsic factor antibodies or antibodies to gastric parietal cells in the setting of symptomatic vitamin B12 deficiency (45); these antibodies are specific but not very sensitive. Therefore, the absence of such antibodies does not exclude the diagnosis of pernicious anemia.
Gastric parietal cell antibodies can be detected in roughly 90% of those with autoimmune atrophic gastritis, which may progress to pernicious anemia as the number of gastric parietal cells decreases. These antibodies are present in approximately 80% to 90% of patients with pernicious anemia in the early stages of the disease. However, as the disease progresses, these antibodies decrease in parallel with the progressive loss of gastric parietal cells, so only about half of the people with advanced pernicious anemia have detectable gastric parietal cell antibodies. Therefore, the absence of gastric parietal cell antibodies doesn’t exclude pernicious anemia. Gastric parietal cell antibodies are also not entirely specific to pernicious anemia, as they can be detected in other autoimmune disorders (eg, Hashimoto thyroiditis, type 1 diabetes). Low levels of these antibodies can also be found in about 5% of healthy people.
Unlike gastric parietal cell antibodies, antibodies against intrinsic factor are moderately sensitive but highly specific for pernicious anemia, although they can rarely exist in immunological diseases. Because only 50% to 70% of patients with pernicious anemia have intrinsic factor antibodies, the lack of these antibodies cannot exclude pernicious anemia; they are, nevertheless, so specific to pernicious anemia that their presence effectively confirms the diagnosis. There are two types of intrinsic factor antibodies: (1) blocking antibodies inhibit cobalamin from binding to intrinsic factor, blocking the formation of the "cobalamin-intrinsic factor" complex that is meant to transport cobalamin safely to the small intestine; and (2) binding antibodies bind to the "cobalamin-intrinsic factor" complex and prevent its absorption at the terminal ileum. Binding antibodies exist almost exclusively in those with blocking antibodies, which is why measurement of blocking antibodies is generally the preferred approach for a diagnostic test.
Gastrin testing. Elevated levels of serum gastrin as a consequence of gastric achlorhydria are characteristic of pernicious anemia, and in patients with low serum B12 levels, serum gastrin assays may be useful in identifying those in whom vitamin B12 deficiency is caused by pernicious anemia (230; 238). However, atrophic gastritis and pernicious anemia in patients with type 1 autoimmune polyglandular syndrome usually demonstrate low or normal gastrin levels, probably because of the loss of gastrin-producing cells (130).
Other testing. In the absence of gastric parietal cell or intrinsic factor antibodies, gastroenterology consultation and other diagnostic tests (eg, endoscopy) may be necessary to determine the cause of cobalamin deficiency if a clear and sufficient explanation is not readily evident from the patient's history (eg, a long history of adherence to a vegan diet, nitrous oxide exposure in surgery or recreationally, history of fish tapeworm infection, or a history of bariatric surgery, gastrectomy, or surgical resection of the terminal ileum). Some other exposures (eg, use of proton pump inhibitors, H2-receptor blocking agents, or metformin) might raise suspicion that this was a significant or sufficient cause, but by itself is not sufficient proof that such an exposure actually caused a vitamin B insufficiency.
• In practice, vitamin B12 supplementation is the appropriate treatment for virtually all forms of vitamin B12 deficiency. | |
• Even with prompt treatment with sufficient doses of cobalamin, the neurologic deficits caused by vitamin B12 deficiency are not always reversible. | |
• If vitamin B12 deficiency is considered a possible cause of existing neurologic deficits, vitamin B12 replacement therapy should be initiated as soon as possible after blood has been taken for diagnostic tests. | |
• Oral vitamin B12 replacement is inexpensive and substantially less costly and more convenient for most patients than parenteral therapy. | |
• A Cochrane review of oral versus intramuscular vitamin B12 concluded that oral and intramuscular administration of vitamin B12 were similarly efficacious, whereas another systematic review in children found that the parenteral route is more efficacious than the oral route, although the quality of available supporting evidence was low for both reviews. | |
• Until better data are available, it is prudent to initiate therapy with at least one intramuscular injection of 1000 µg. This could then be followed by several weeks of very high-dose oral therapy (2 mg cobalamin daily). Thereafter, daily doses of 1 mg of cobalamin should maintain adequate body B12 stores, even in cases of pernicious anemia. | |
• Whether oral or parenteral maintenance therapy is chosen, it is crucial that efficacy is monitored initially by clinical improvement. | |
• Sustained correction of vitamin B12 deficiency should be checked annually by serum vitamin B12, methylmalonic acid, or total homocysteine levels. | |
• Pernicious anemia requires lifelong treatment, and studies have shown substantial rates of noncompliance with vitamin B12 supplementation. |
In practice, vitamin B12 supplementation is the appropriate treatment for virtually all forms of vitamin B12 deficiency. The optimal dosage and route of administration for cobalamin repletion and ongoing supplementation have been somewhat controversial because of inadequate studies establishing an optimal treatment regimen for affected individuals, and particularly for those with neurologic manifestations (198). In addition, diagnostic thresholds developed for adults may not be appropriate for children and adolescents (198).
Some of the non-neurologic manifestations of vitamin B12 deficiency, such as glossitis, respond promptly to vitamin B12 repletion (277).
Even with prompt treatment with sufficient doses of cobalamin, the neurologic deficits caused by vitamin B12 deficiency are not always reversible. Therefore, if vitamin B12 deficiency is considered a possible cause of existing neurologic deficits, vitamin B12 replacement therapy should be initiated as soon as possible after blood has been taken for diagnostic tests. Traditionally, this initial therapy has consisted of four to seven daily parenteral (usually intramuscular) injections of 1 mg of cobalamin or an intramuscular injection of 1 mg of vitamin B12 at weekly intervals for 1 month (238). The reasoning has been that vitamin B12 stores can be repleted faster and more surely by parenteral administration than by the oral route. This initial therapy is then traditionally followed by monthly injections of 1 mg of cobalamin.
The pharmacologic superiority of parenteral vitamin B12 to oral supplementation with large doses of cobalamin has never been adequately demonstrated.
In the absence of intrinsic factor (eg, from pernicious anemia or gastrectomy), the gut absorbs about 1% of ingested cobalamin, and this appears to be true even in patients with diseases of the ileum. In studies comparing the effect of oral and parenteral cobalamin, large doses (2 to 3 mg) of daily oral B12 appear equally effective to daily parental cobalamin 1 mg in rapidly restoring cobalamin stores and treating the signs of deficiency (215; 136; 40). Even in patients with inflammatory bowel disease affecting the terminal ileum (eg, Crohn disease), oral supplementation of vitamin B12 appears to be effective (84). In children under 2 years of age, an oral dose of 1 mg daily is effective, but 2 mg daily is recommended for repleting children who are older than 6 years of age (25). Oral vitamin B12 replacement is inexpensive and substantially less costly and more convenient for most patients than parenteral therapy (32; 165).
A Cochrane review concluded that oral and intramuscular administration of vitamin B12 were similarly efficacious; however, the quality of available supporting evidence was low (269). This was also true of a subsequent meta-analysis (01).
Another systematic review in children, based on limited evidence from a single, methodologically weak randomized controlled trial, suggested that the parenteral route is more efficacious than the oral route (218).
Further, none of the studies cited above included large numbers of patients with neurologic deficits, although significant improvement occurred in the limited number of orally treated patients with such deficits. The neurologic deficits included in these studies tended toward mild sensory phenomena rather than severe myelopathic manifestations. It has specifically not been shown that oral vitamin B12 therapy is as good as parenteral therapy to alleviate neurologic symptoms, particularly in subjects with severe neurologic presentations (274).
Therefore, until better data are available, it is prudent to initiate therapy with at least one intramuscular injection of 1 mg. This could then be followed by one of the following: (1) three to six daily intramuscular injections of 1 mg of cobalamin, (2) intramuscular injection of 1 mg vitamin B12 at weekly intervals for 1 month, or (3) several weeks of very high-dose oral therapy (2 mg cobalamin daily). Until more definitive studies are available, severe neurologic presentations should still receive a longer course of parenteral vitamin B12 repletion. Thereafter, daily doses of 1 mg of cobalamin should maintain adequate body B12 stores, even in cases of pernicious anemia.
Sublingual vitamin B12 preparations are increasingly common and are likely as effective as traditional oral supplementation (37).
Whether oral or parenteral maintenance therapy is chosen, it is crucial that efficacy is monitored initially by clinical improvement. Sustained correction of vitamin B12 deficiency should be checked annually by serum vitamin B12, methylmalonic acid, or total homocysteine levels (46). Pernicious anemia requires lifelong treatment, and studies have shown substantial rates of noncompliance with vitamin B12 supplementation (45).
Maternal vitamin B12 deficiency or borderline deficiency presents two major risks to the developing fetus and infant. First, vitamin B12 deficiency may increase the risk of neural tube defects (128; 208; 251; 22). Second, neurologic and hematologic manifestations of vitamin B12 deficiency may occur in infants breastfed by mothers who have subnormal stores of cobalamin (86; 99; 244; 119). The placenta avidly procures cobalamin for the fetus; consequently, deficiency does not usually manifest until infancy, usually within the first year. Vitamin B12 deficiency in infants may cause irreversible developmental impairment. Infants of mothers on vegan or vegetarian diets are at significant risk of vitamin B12 deficiency unless the mother takes supplementary vitamin B12 (119).
Women with preeclampsia have significantly lower vitamin B12 concentrations than normotensive pregnant women (161).
The anesthetic agent nitrous oxide irreversibly inactivates cobalamin. Typical neurologic symptoms of vitamin B12 deficiency may develop over a few days or weeks after a single exposure to nitrous oxide in patients with borderline or deficient stores of vitamin B12 (88). This condition should be promptly treated with parenteral vitamin B12 or permanent neurologic damage may occur. Symptoms of vitamin B12 deficiency will also develop in some who chronically use nitrous oxide as a recreational drug, even in the absence of vitamin B12 deficiency (143).
Nitrous oxide dose during labor is a predictor for total homocysteine and may impact the interpretation of total homocysteine analysis in newborn screening (154). In addition, nitrous oxide administration during labor may be a contributing risk factor for infants prone to developing vitamin B12 deficiency (154).
Nitrous oxide-mediated vitamin B12 deficiency may lead to an acute prothrombotic state by elevating homocysteine levels (23). Ninety patients undergoing endarterectomy were randomized to anesthesia with or without nitrous oxide. Postoperative homocysteine levels were higher in the patients randomized to nitrous oxide anesthesia, and ischemic signs on the ECG were significantly more frequent in this group.
Preoperative vitamin supplementation with vitamin B12 can prevent the nitrous oxide-induced elevation in homocysteine levels (24; 127).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Douglas J Lanska MD MS MSPH
Dr. Lanska of the University of Wisconsin School of Medicine and Public Health has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neurobehavioral & Cognitive Disorders
Jun. 17, 2026
Neuro-Oncology
May. 27, 2026
Neuropharmacology & Neurotherapeutics
May. 14, 2026
Neuro-Oncology
Apr. 30, 2026
Neuropharmacology & Neurotherapeutics
Apr. 23, 2026
Neuropharmacology & Neurotherapeutics
Apr. 20, 2026
Neuro-Ophthalmology & Neuro-Otology
Apr. 07, 2026
General Neurology
Apr. 06, 2026